A Diagnostic Case Study for Manufacturing Gas-Phase Chemical Sensors

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
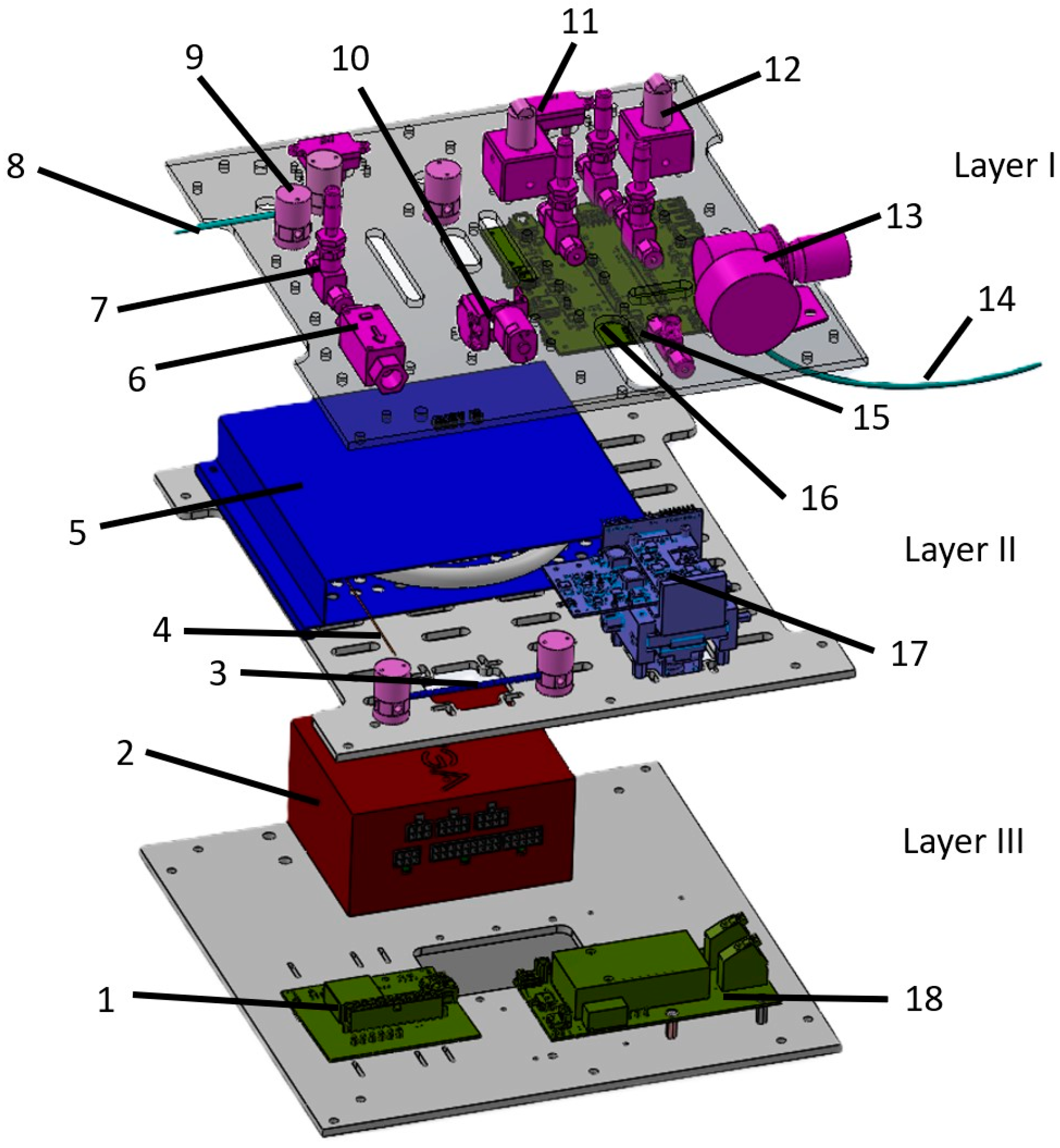
2.1. System Overview of the Device
2.2. Case Presentation—Device Components
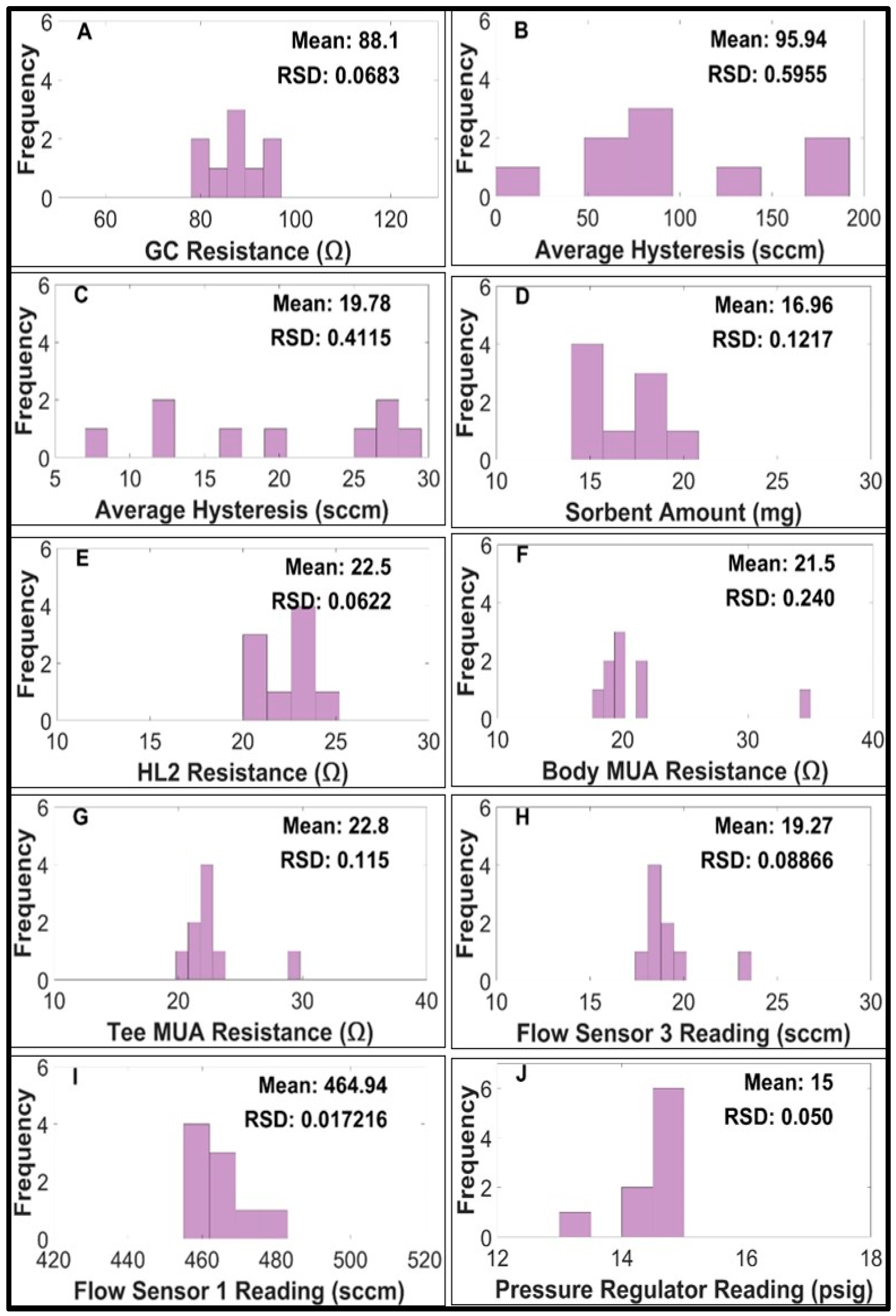
2.2.1. Performance of the Gas Chromatography Columns
2.2.2. Proportional Valves
2.2.3. Sorbent Trap
2.2.4. Heated Transfer Lines
2.2.5. Flow Sensors
2.2.6. Pressure Regulator
2.2.7. Ionization Source
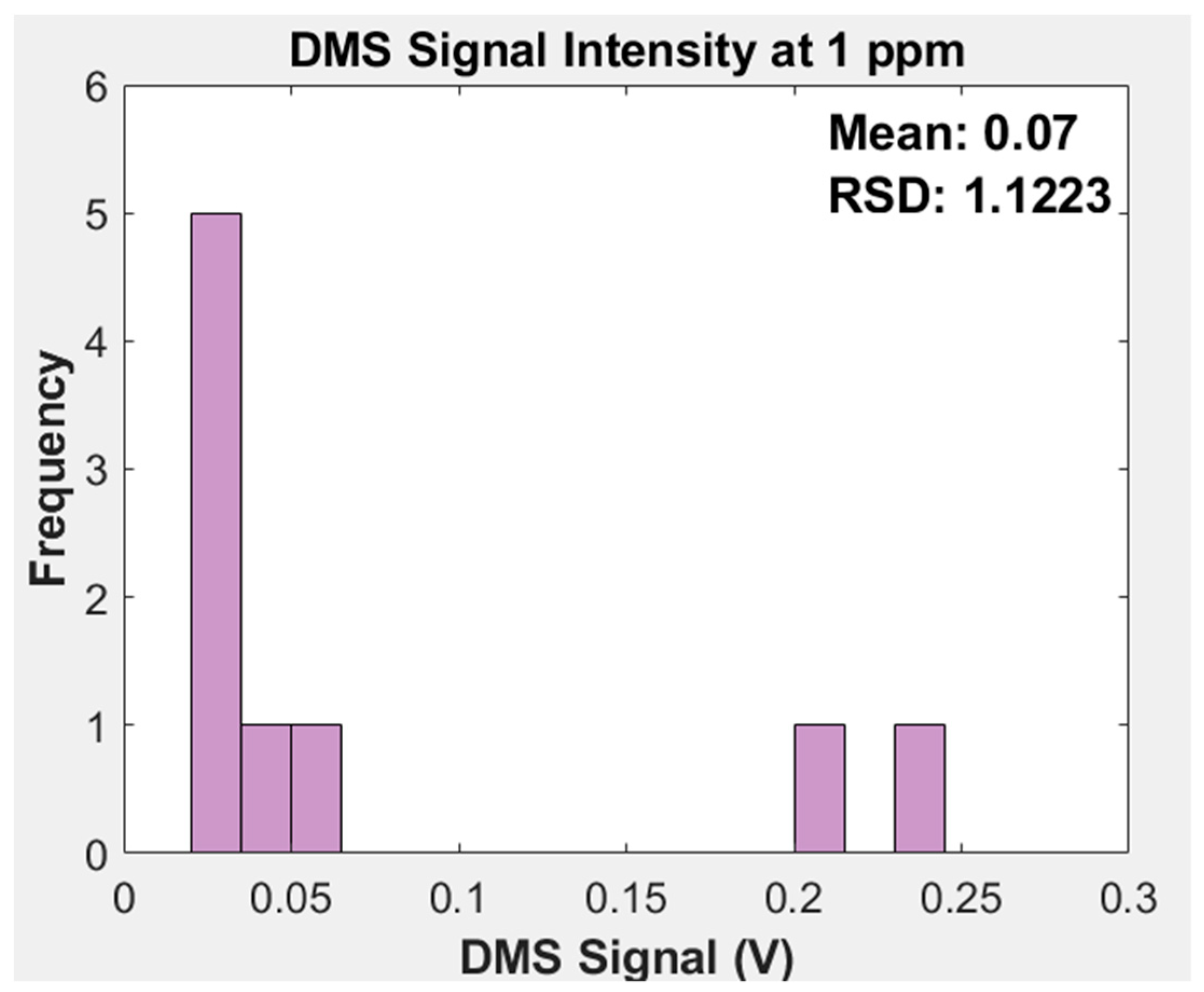
2.2.8. Differential Mobility Spectrometer
3. Results
3.1. Device Failure Modes
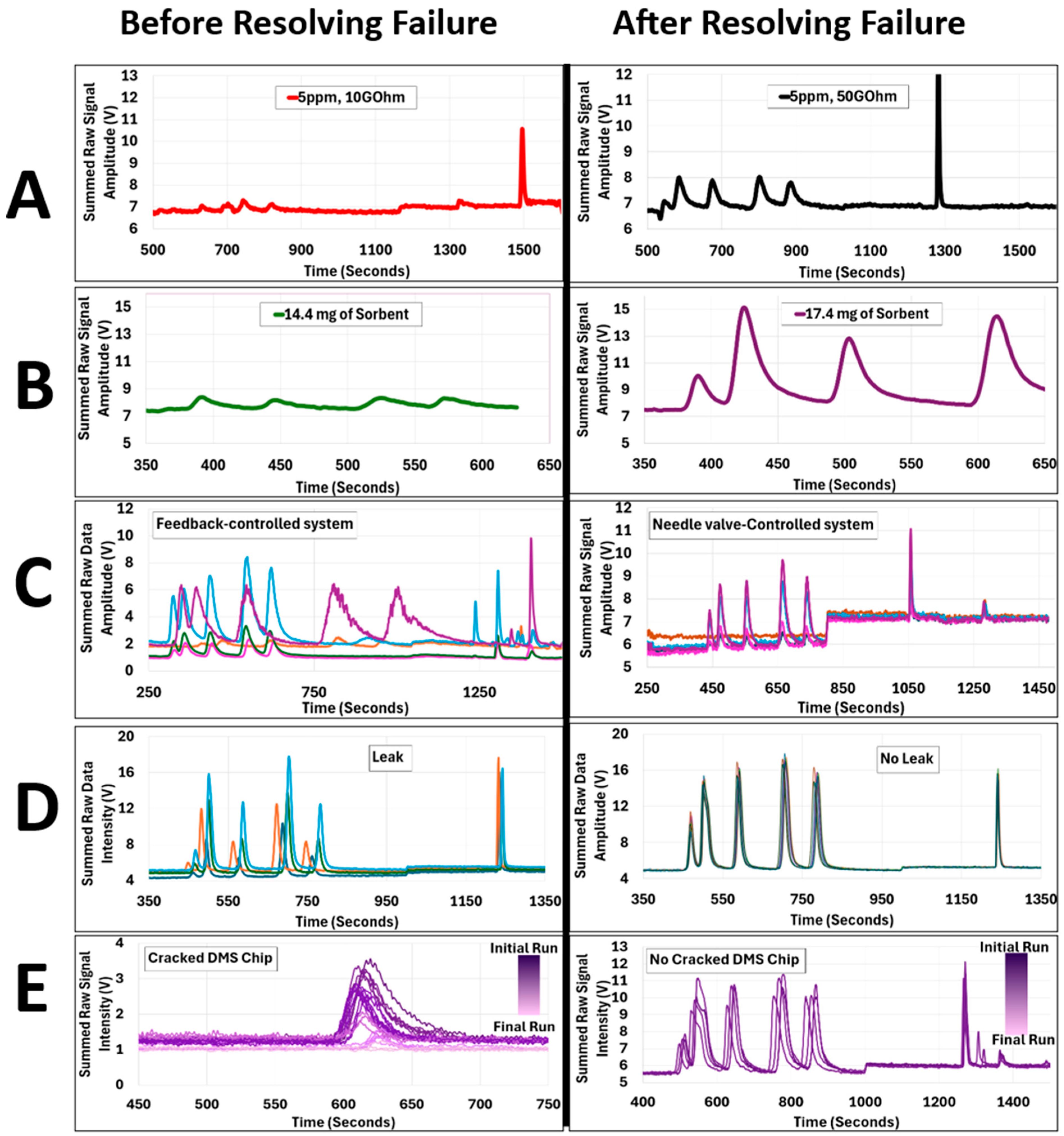
3.1.1. Failure Mode A: Gain Resistor
3.1.2. Failure Mode B: Sorbent Trap
3.1.3. Failure Mode C: Feedback Controlled System Failure Resulting from the Inherent Inaccuracies of Flow Sensors and the Hysteresis of the Proportional Control Valve
3.1.4. Failure Mode D: Leaks
3.1.5. Failure Mode E: DMS Chip Failure
3.2. Management and Outcome
4. Discussion
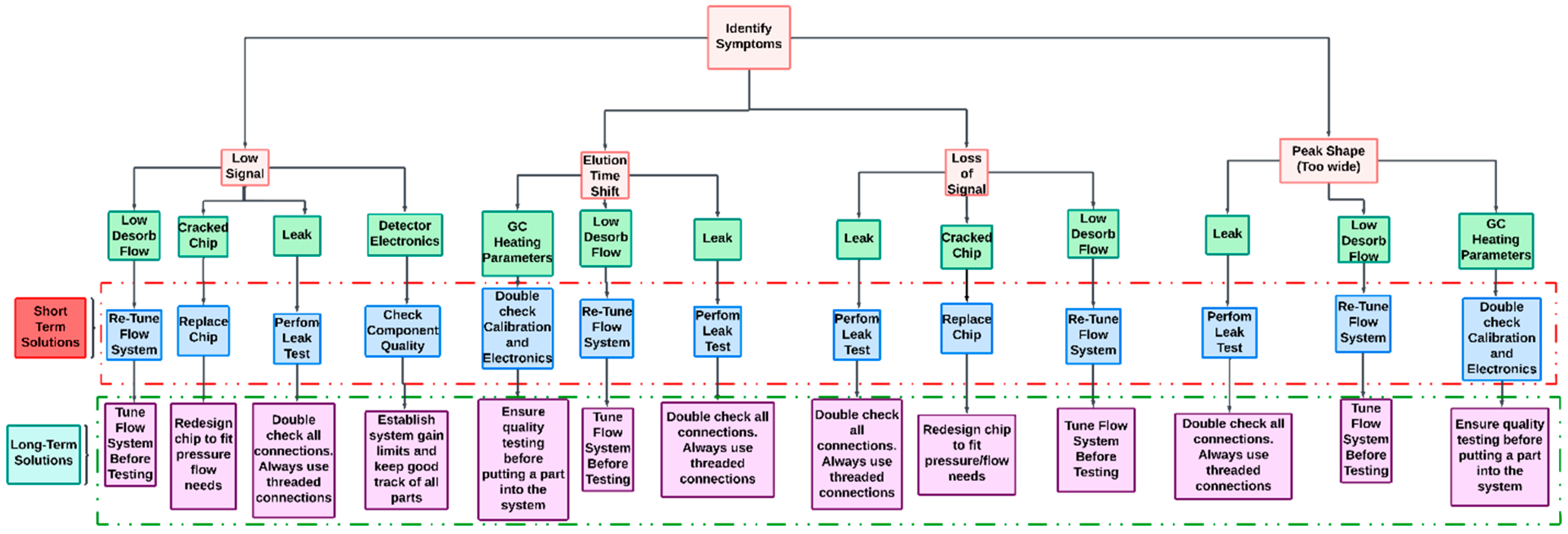
4.1. Symptom: Low Signal
4.2. Symptom: Elution Time Shifts
4.3. Symptom: Loss of Signal
4.4. Symptom: Peak Shape/Width Variation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabianowski, W.; Maziejuk, M.; Szyposzyńska, M.; Wiśnik-Sawka, M. Detection and identification of vocs using differential ion mobility spectrometry (Dms). Molecules 2021, 27, 234. [Google Scholar] [CrossRef]
- Frausto-Vicencio, I.; Moreno, A.; Goldsmith, H.; Hsu, Y.-K.; Hopkins, F.M. Characterizing the performance of a compact btex gc-pid for near-real time analysis and field deployment. Sensors 2021, 21, 2095. [Google Scholar] [CrossRef] [PubMed]
- Guntner, A.T.; Abegg, S.; Konigstein, K.; Gerber, P.A.; Schmidt-Trucksass, A.; Pratsinis, S.E. Breath sensors for health monitoring. ACS Sens. 2019, 4, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Camara, M.; Gharbi, N.; Lenouvel, A.; Behr, M.; Guignard, C.; Orlewski, P.; Evers, D. Detection and quantification of natural contaminants of wine by gas chromatography–differential ion mobility spectrometry (GC-DMS). J. Agric. Food Chem. 2013, 61, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Buryakov, I.; Krylov, E.; Nazarov, E.; Rasulev, U.K. A new method of separation of multi-atomic ions by mobility at atmospheric pressure using a high-frequency amplitude-asymmetric strong electric field. Int. J. Mass Spectrom. Ion Process. 1993, 128, 143–148. [Google Scholar] [CrossRef]
- Bílek, J.; Maršolek, P.; Bílek, O.; Buček, P. Field test of mini photoionization detector-based sensors—Monitoring of volatile organic pollutants in ambient air. Environments 2022, 9, 49. [Google Scholar] [CrossRef]
- Huang, X.; Yang, S.; Li, W.; Nidetz, R.; Sharma, R.; Sivakumar, A.D.; Thota, C.; Zhu, H.; Wu, W.; Jeong, S.-Y. Microfluidic integration of μPID on μcolumn for ultracompact micro-gas chromatography. Sens. Actuators B Chem. 2024, 410, 135717. [Google Scholar] [CrossRef]
- Agbroko, S.O.; Covington, J. A novel, low-cost, portable PID sensor for the detection of volatile organic compounds. Sens. Actuators B Chem. 2018, 275, 10–15. [Google Scholar] [CrossRef]
- Tereshkov, M.; Dontsova, T.; Saruhan, B.; Krüger, S. Metal Oxide-Based Sensors for Ecological Monitoring: Progress and Perspectives. Chemosensors 2024, 12, 42. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Li, J.; Duan, Z.; Yang, Y.; Yuan, Z.; Jiang, Y.; Tai, H. Pd-decorated ZnO hexagonal microdiscs for NH3 sensor. Chemosensors 2024, 12, 43. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, P.; Cao, W.; Li, X.; Lai, J. High-sensitivity ethylene gas sensor based on NDIR and dual-channel lock-in amplifier. Optik 2020, 223, 165630. [Google Scholar] [CrossRef]
- Xu, M.; Peng, B.; Zhu, X.; Guo, Y. Multi-gas detection system based on non-dispersive infrared (NDIR) spectral technology. Sensors 2022, 22, 836. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.; Contreras, R.P.; Fung, A.G.; Gibson, P.; LeVasseur, M.K.; McCartney, M.M.; Koch, D.T.; Chakraborty, P.; Chew, B.S.; Rajapakse, M.Y. Portable chemical detection platform for on-site monitoring of odorant levels in natural gas. J. Chromatogr. A 2023, 1705, 464151. [Google Scholar] [CrossRef] [PubMed]
- Yeap, D.; Hichwa, P.T.; Rajapakse, M.Y.; Peirano, D.J.; McCartney, M.M.; Kenyon, N.J.; Davis, C.E. Machine vision methods, natural language processing, and machine learning algorithms for automated dispersion plot analysis and chemical identification from complex mixtures. Anal. Chem. 2019, 91, 10509–10517. [Google Scholar] [CrossRef] [PubMed]
- Peirano, D.J.; Pasamontes, A.; Davis, C.E. Supervised semi-automated data analysis software for gas chromatography/differential mobility spectrometry (GC/DMS) metabolomics applications. Int. J. Ion Mobil. Spectrom. 2016, 19, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Rajapakse, M.Y.; McCartney, M.M.; Kenyon, N.J.; Davis, C.E. Machine learning and signal processing assisted differential mobility spectrometry (DMS) data analysis for chemical identification. Anal. Methods 2022, 14, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Chromatotech. energyMEDOR. Available online: https://chromatotec.com/solutions/auto-gc-866-analyzers-range/sulfur-compounds/energymedor/ (accessed on 23 July 2024).
- Miller, R.A.; Nazarov, E.G.; Eiceman, G.A.; King, A.T. A MEMS radio-frequency ion mobility spectrometer for chemical vapor detection. Sens. Actuators A Phys. 2001, 91, 301–312. [Google Scholar] [CrossRef]
- Moretti, P.; Vezzani, S.; Garrone, E.; Castello, G. Evaluation and prediction of the shape of gas chromatographic peaks. J. Chromatogr. A 2004, 1038, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.V.; Bahaghighat, H.D.; Synovec, R.E. Column selection approach to achieve a high peak capacity in comprehensive three-dimensional gas chromatography. Talanta 2019, 195, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Siegler, W.C.; Hoggard, J.C.; Fitz, B.D.; Nadeau, J.S.; Synovec, R.E. Achieving high peak capacity production for gas chromatography and comprehensive two-dimensional gas chromatography by minimizing off-column peak broadening. J. Chromatogr. A 2011, 1218, 3130–3139. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, R.P.; Koch, D.T.; Gibson, P.; McCartney, M.M.; Chew, B.S.; Chakraborty, P.; Chevy, D.A.; Honeycutt, R.; Haun, J.; Griffin, T.; et al. A Diagnostic Case Study for Manufacturing Gas-Phase Chemical Sensors. Chemosensors 2024, 12, 155. https://doi.org/10.3390/chemosensors12080155
Contreras RP, Koch DT, Gibson P, McCartney MM, Chew BS, Chakraborty P, Chevy DA, Honeycutt R, Haun J, Griffin T, et al. A Diagnostic Case Study for Manufacturing Gas-Phase Chemical Sensors. Chemosensors. 2024; 12(8):155. https://doi.org/10.3390/chemosensors12080155
Chicago/Turabian StyleContreras, Raquel Pimentel, Dylan T. Koch, Patrick Gibson, Mitchell M. McCartney, Bradley S. Chew, Pranay Chakraborty, Daniel A. Chevy, Reid Honeycutt, Joseph Haun, Thomas Griffin, and et al. 2024. "A Diagnostic Case Study for Manufacturing Gas-Phase Chemical Sensors" Chemosensors 12, no. 8: 155. https://doi.org/10.3390/chemosensors12080155