
A Deferasirox Derivative That Acts as a Multifaceted Platform for the Detection and Quantification of Fe3+

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Chemicals
2.3. Water Samples
2.4. pH Determination
2.5. Spectroscopy
2.6. Cell Experiments
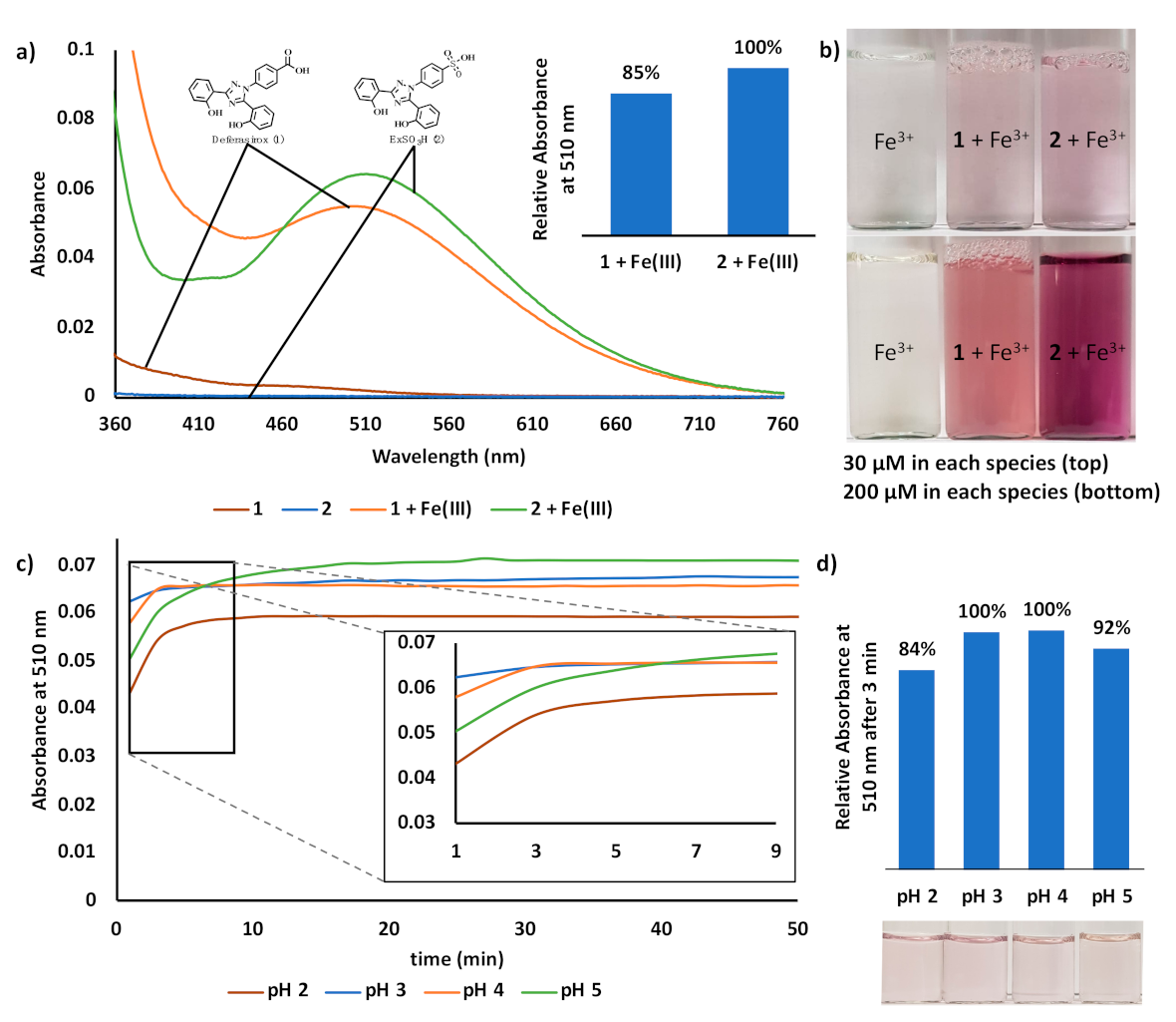
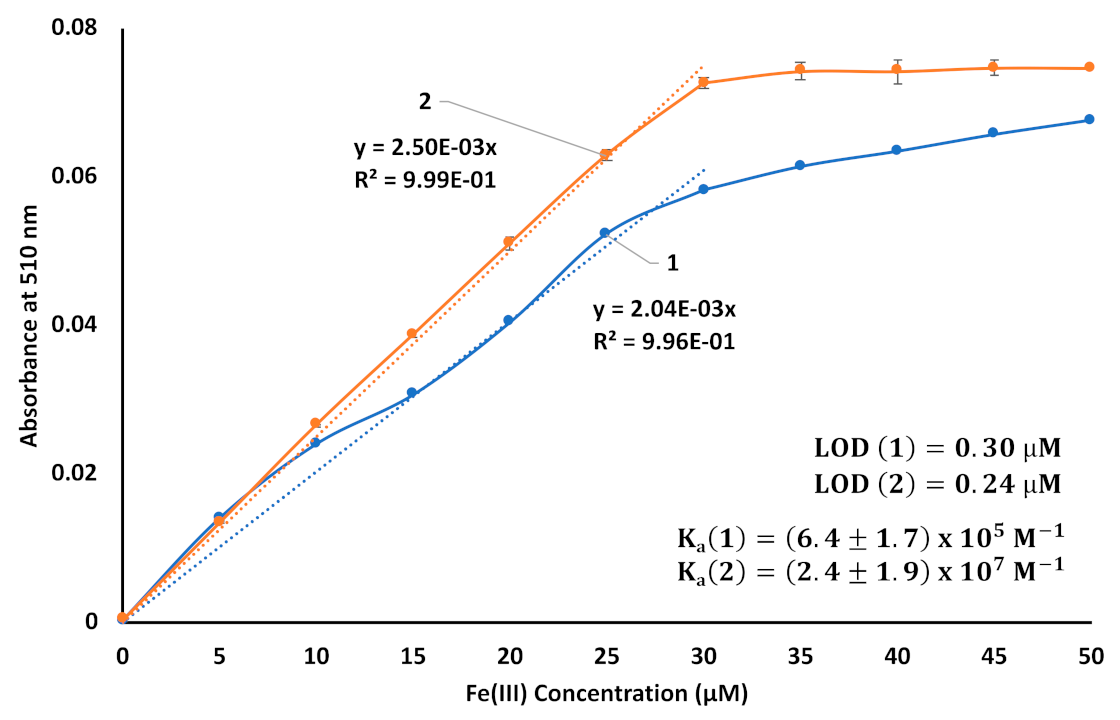
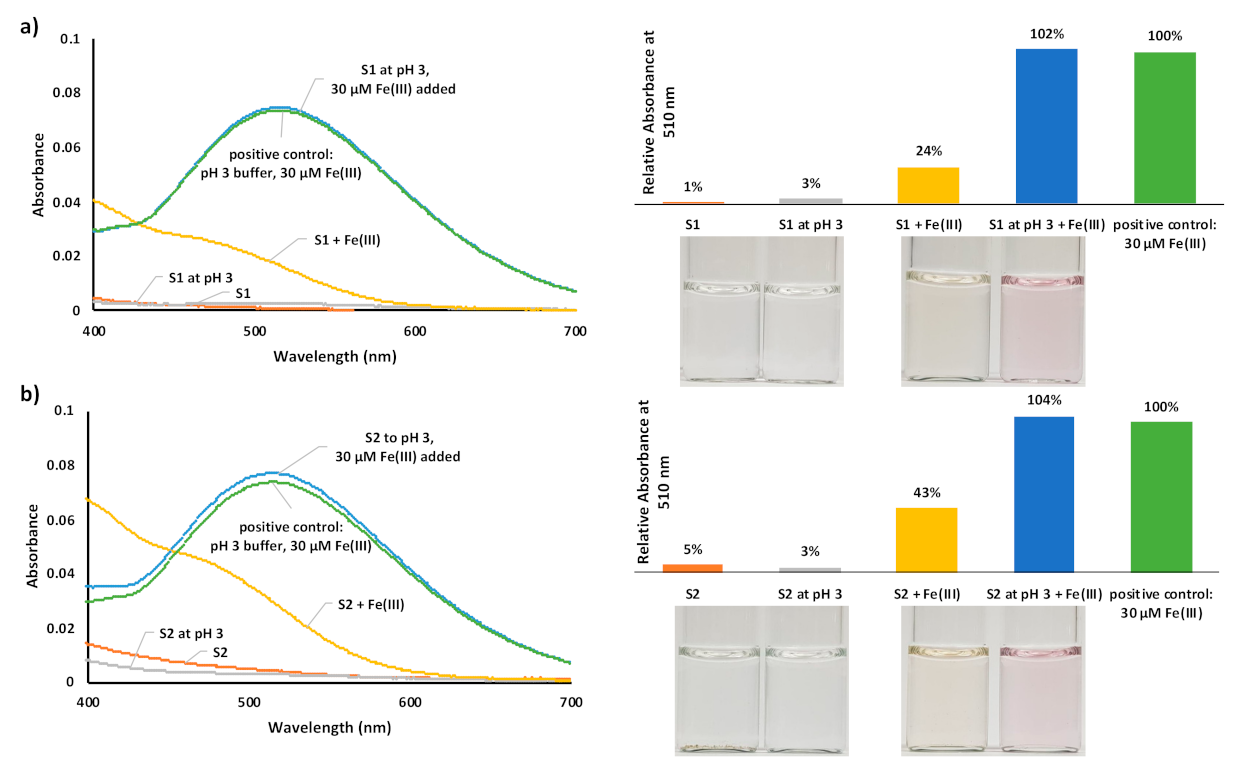
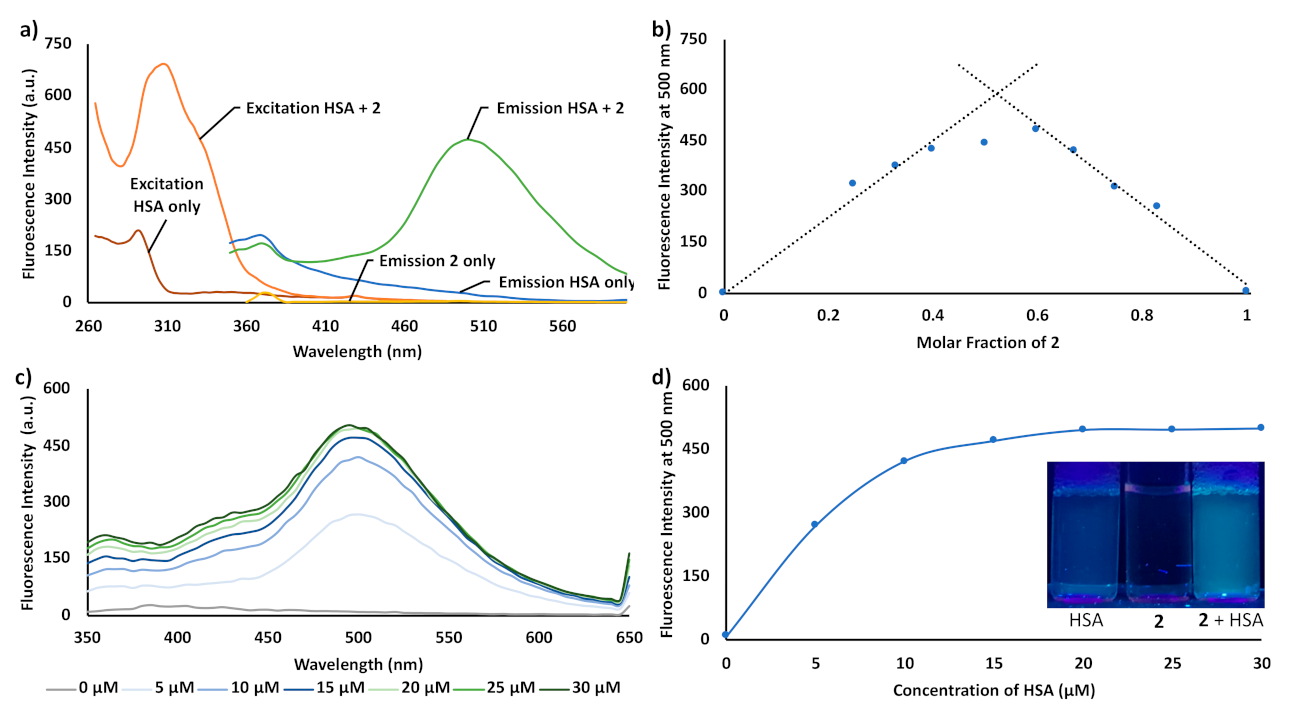
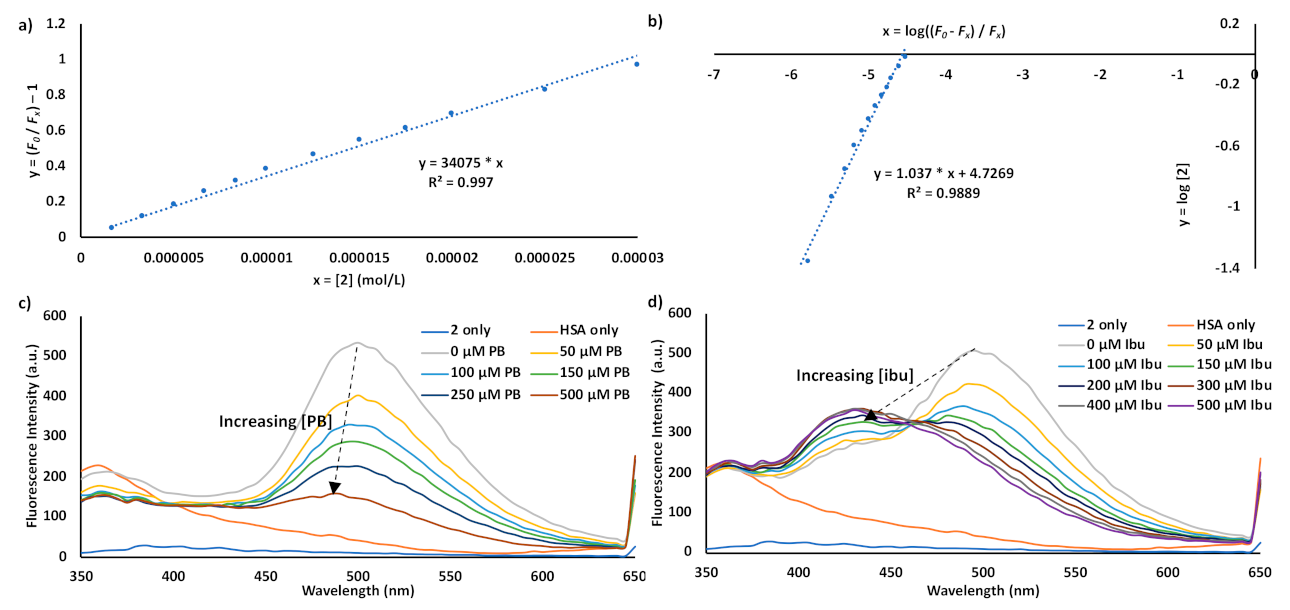
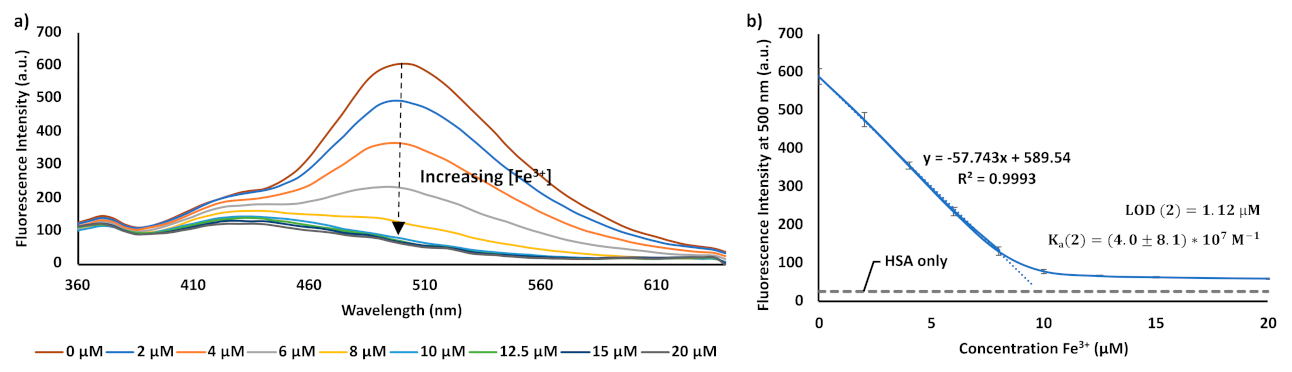
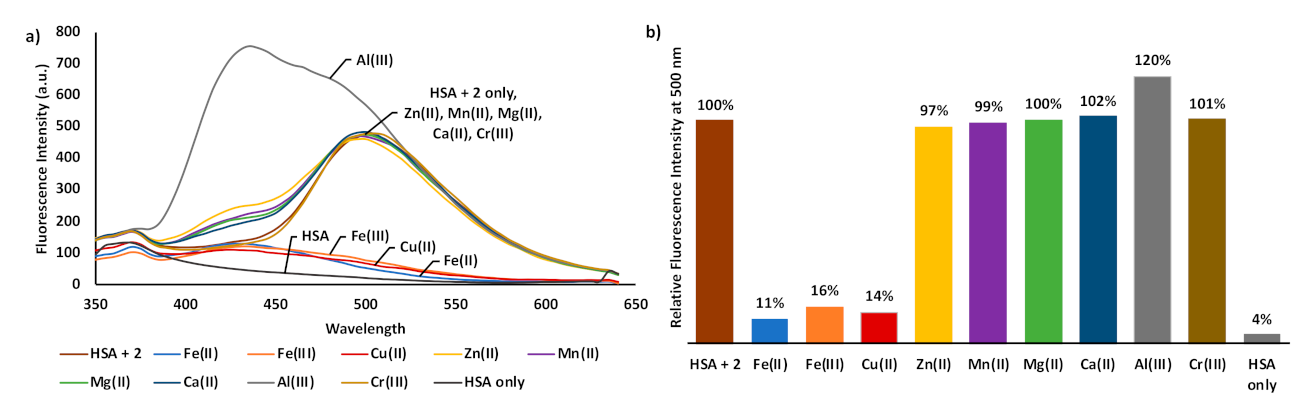
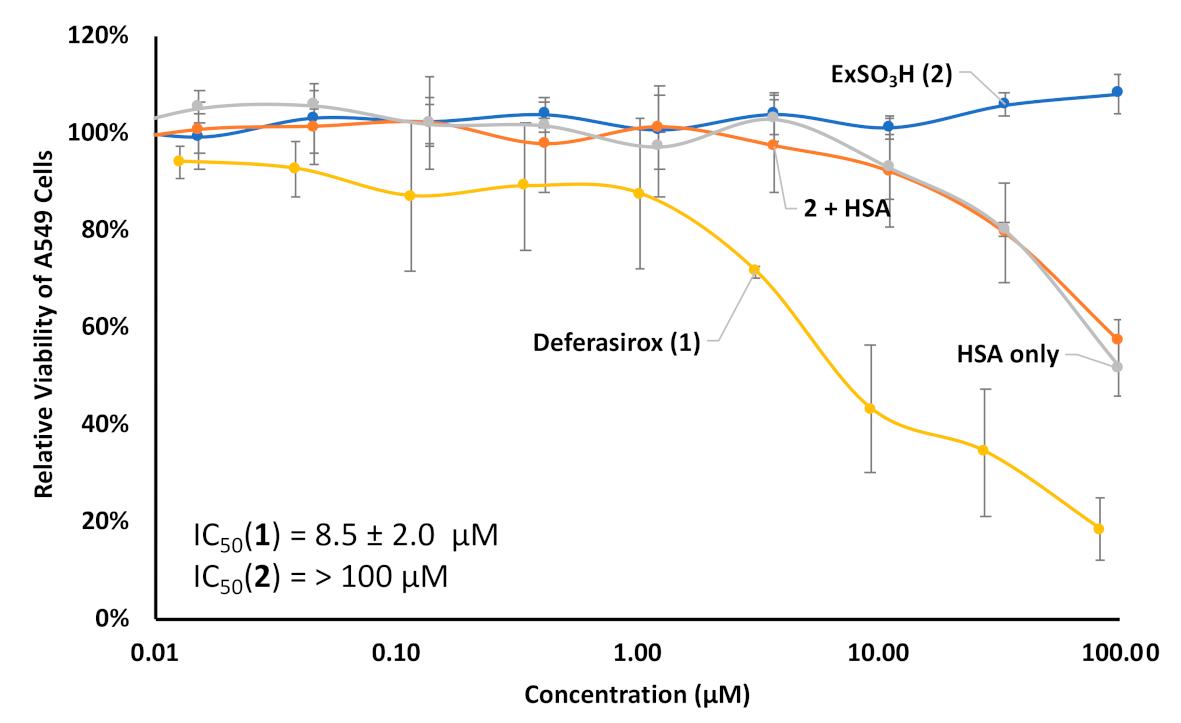
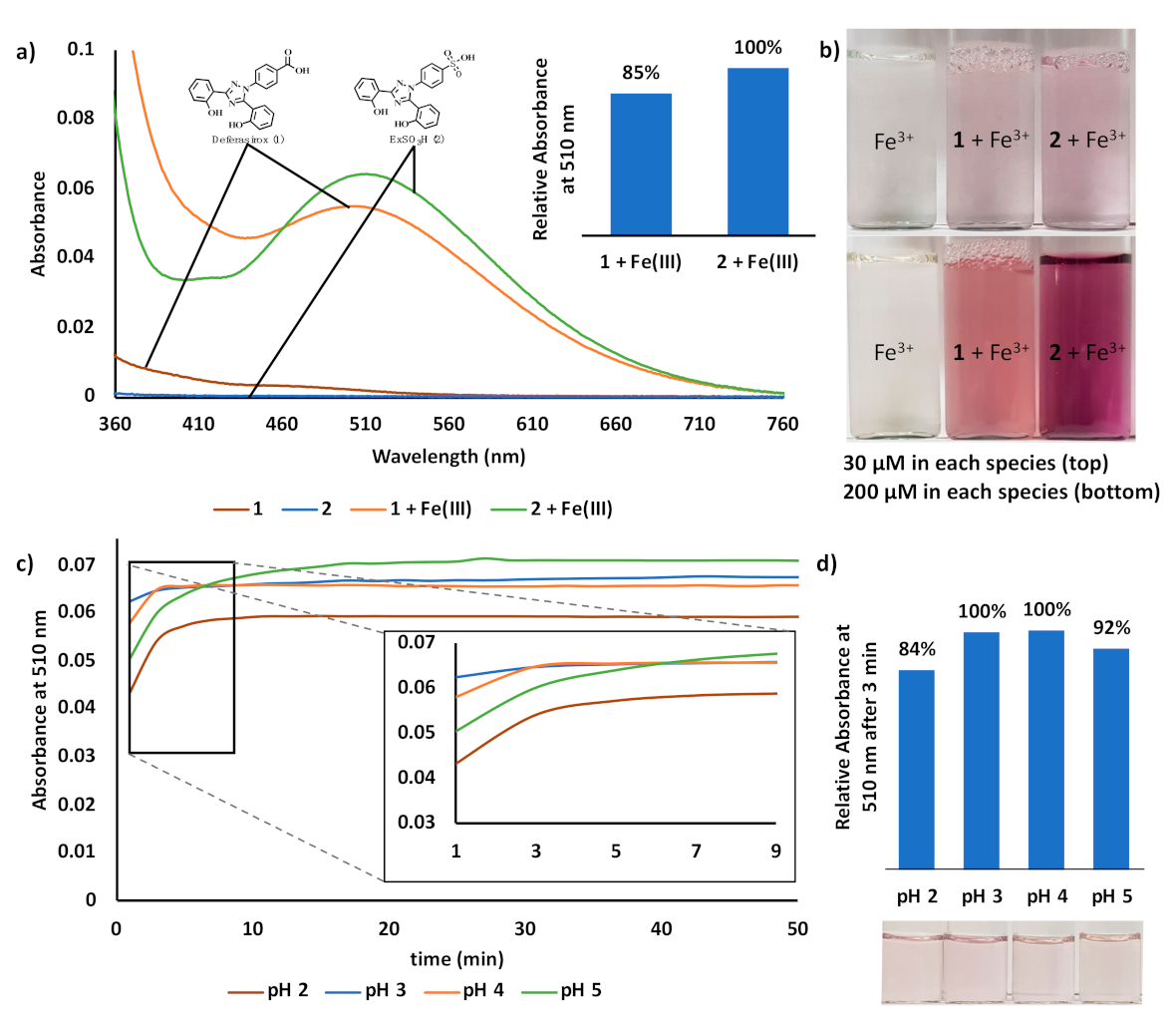
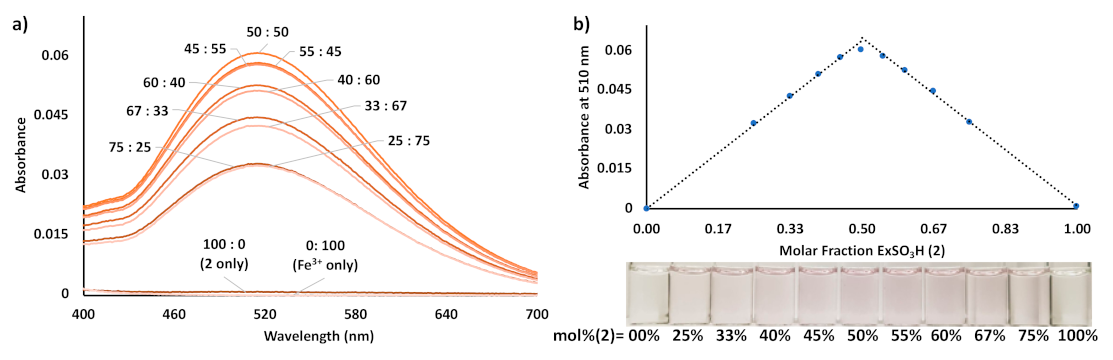
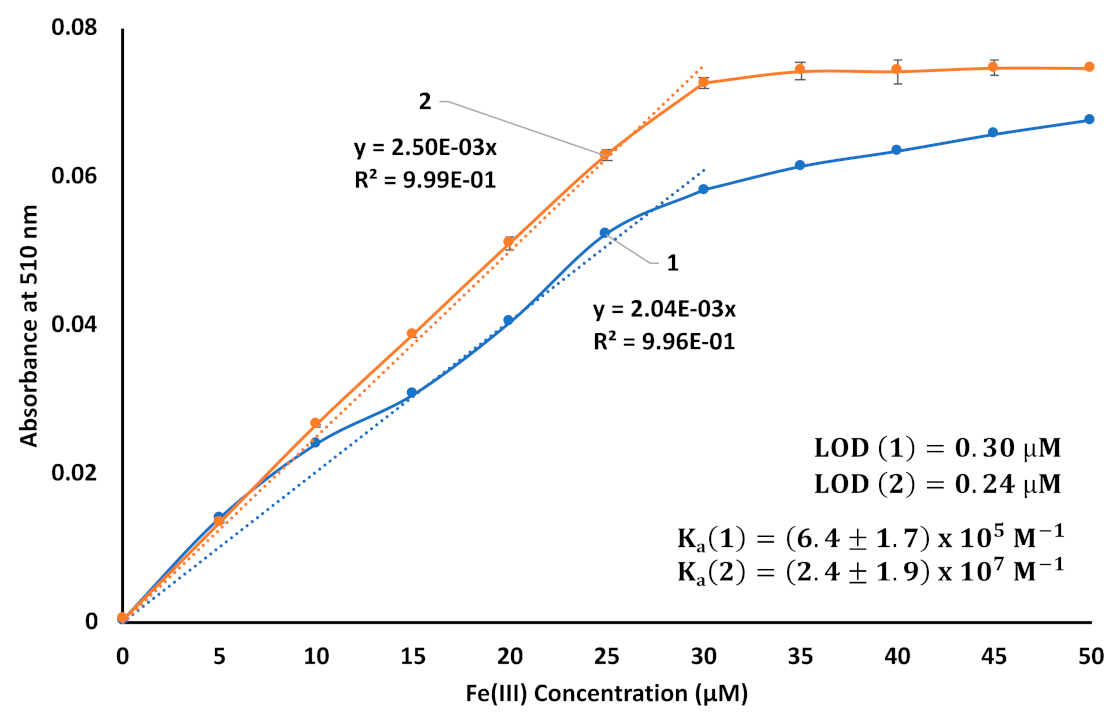
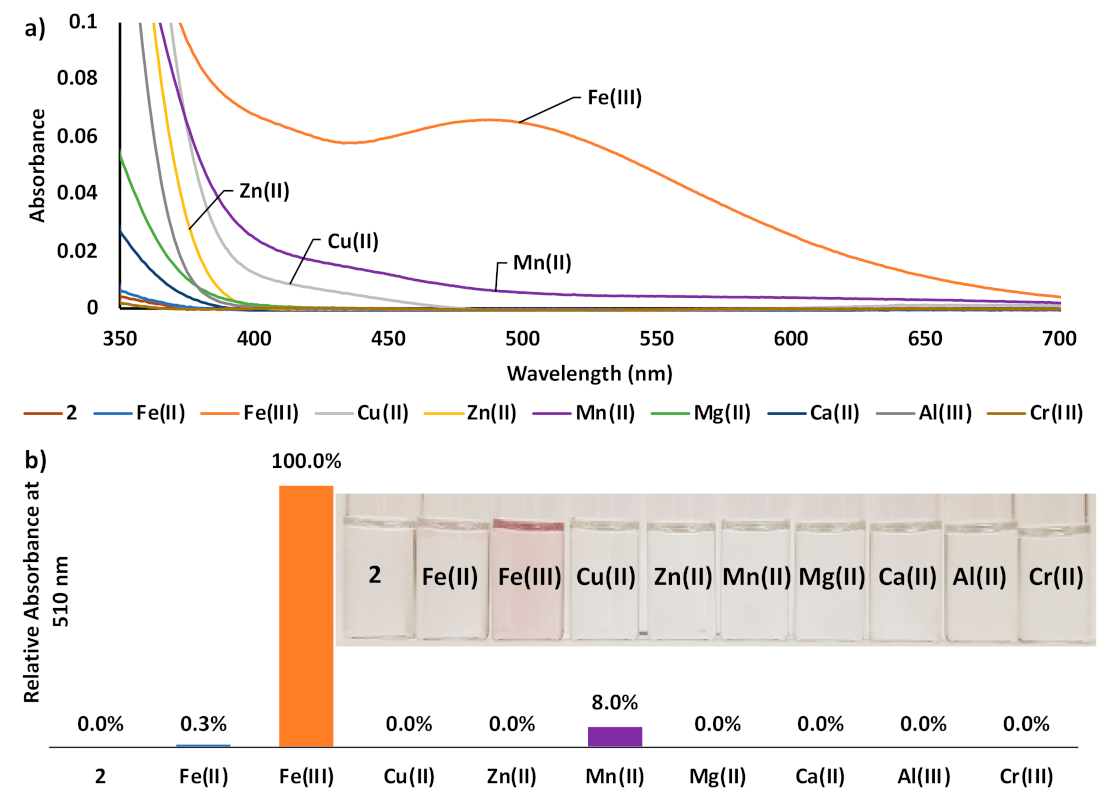
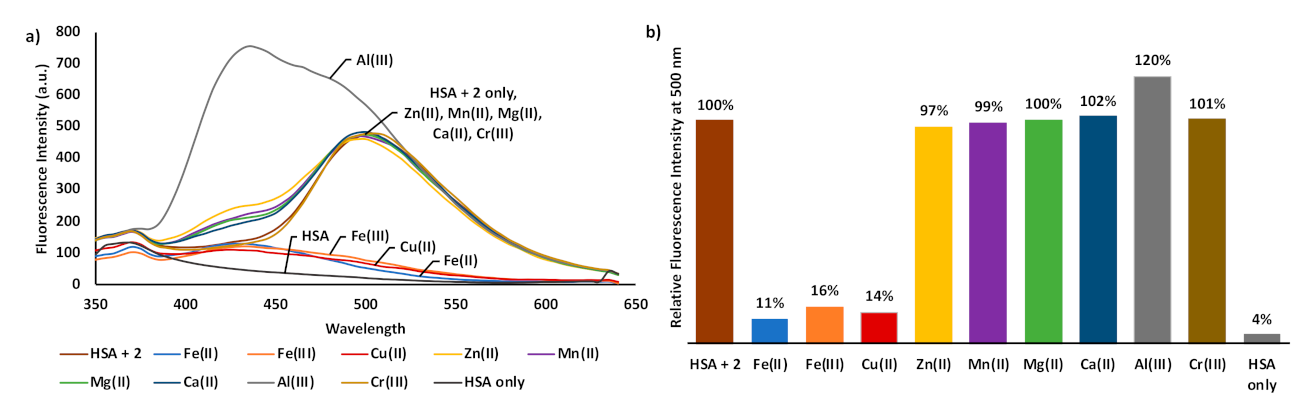
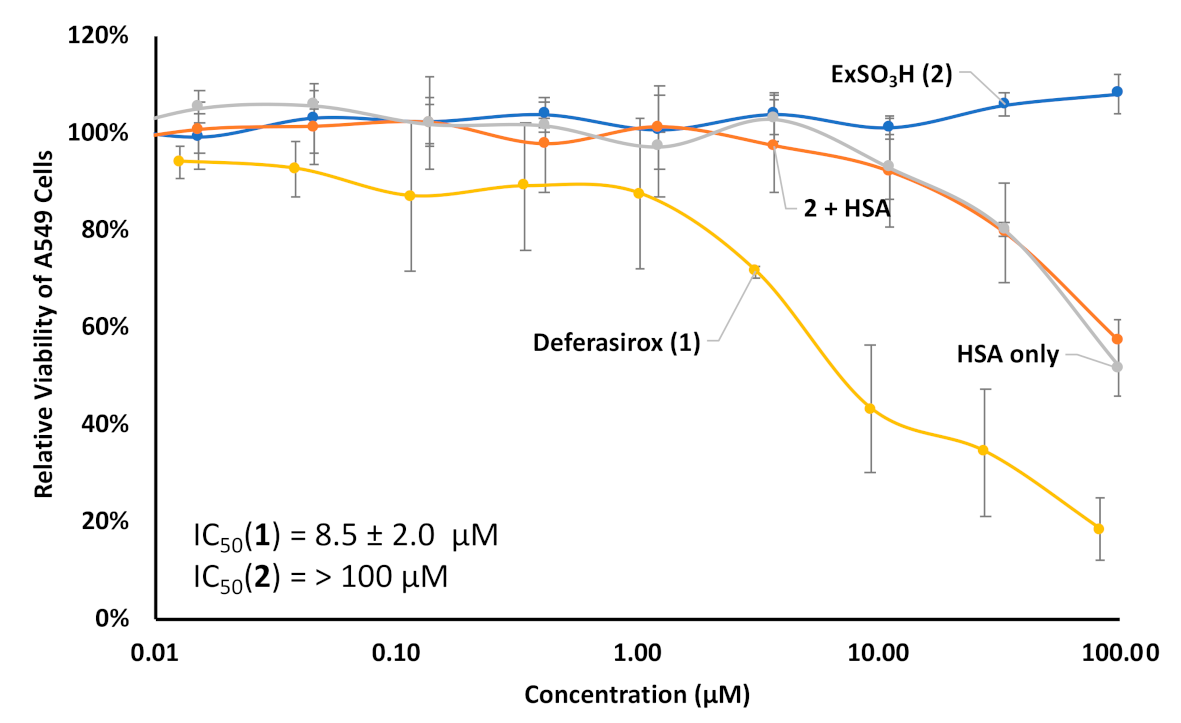
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization; International Programme on Chemical Safety. Guidelines for Drinking-Water Quality: Health Criteria and Other Supporting Information, 2nd ed.; WHO: Geneva, Switzerland, 1996; Volume 2. [Google Scholar]
- Pesavento, M.; Alberti, G.; Biesuz, R. Analytical methods for determination of free metal ion concentration, labile species fraction and metal complexation capacity of environmental waters: A review. Anal. Chim. Acta 2009, 631, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in Neurobiology: Probing Their Chemistry and Biology with Molecular Imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef] [PubMed]
- Steinbrueck, A.; Sedgwick, A.C.; Brewster, J.T.; Yan, K.-C.; Shang, Y.; Knoll, D.M.; Vargas-Zúñiga, G.I.; He, X.-P.; Tian, H.; Sessler, J.L. Transition metal chelators, pro-chelators, and ionophores as small molecule cancer chemotherapeutic agents. Chem. Soc. Rev. 2020, 49, 3726–3747. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 702–717. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Fujii, M.; Imaoka, A.; Yoshimura, C.; Waite, T.D. Effects of molecular composition of natural organic matter on ferric iron complexation at circumneutral pH. Environ. Sci. Technol. 2014, 48, 4414–4424. [Google Scholar] [CrossRef]
- Schwertmann, U. Solubility and dissolution of iron oxides. Plant Soil 1991, 130, 1–25. [Google Scholar] [CrossRef]
- Arpadjan, S.; Tsekova, K.; Petrova, P.; Knutsson, J. Field sampling, speciation and determination of dissolved iron (II) and iron (III) in waters. Bulg. Chem. Commun. 2012, 44, 299–306. [Google Scholar] [CrossRef]
- Vickackaite, V.; Tautkus, S.; Kazlauskas, R. Determination of Heavy Metals in Natural Waters by Flame Atomic Absorption Spectrometry. Chem. Anal. 1996, 41, 483–488. [Google Scholar]
- Shamspur, T.; Sheikhshoaie, I.; Mashhadizadeh, M.H. Flame atomic absorption spectroscopy (FAAS) determination of iron(III) after preconcentration on to modified analcime zeolite with 5-((4-nitrophenylazo)-N-(2′,4′-dimethoxyphenyl))salicylaldimine by column method. J. Anal. At. Spectrom. 2005, 20, 476–478. [Google Scholar] [CrossRef]
- Yildiz, O.; Citak, D.; Tuzen, M.; Soylak, M. Determination of copper, lead and iron in water and food samples after column solid phase extraction using 1-phenylthiosemicarbazide on Dowex Optipore L-493 resin. Food Chem. Toxicol. 2011, 49, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.P.; Hendry, J.M.; Kerrich, R. Speciation of dissolved iron(III) and iron(II) in water by on-line coupling of flow injection separation and preconcentration with inductively coupled plasma mass spectrometry. Anal. Chem. 2000, 72, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q. Simultaneous Separation and Quantification of Iron and Transition Species Using LC-ICP-MS. Am. J. Anal. Chem. 2011, 2, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Pomazal, K.; Prohaska, C.; Steffan, I.; Reich, G.; Huber, J.F.K. Determination of Cu, Fe, Mn, and Zn in blood fractions by SEC-HPLC-ICP-AES coupling. Analyst 1999, 124, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Rouge, P.; Gervais, C.; Courty, M.; Dassonville-Klimpt, A.; Sonnet, P.; Baudrin, E. A new sensitive organic/inorganic hybrid material based on titanium oxide for the potentiometric detection of iron(III). J. Colloid Interface Sci. 2012, 388, 130–136. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.M.G. Chemical speciation of iron in seawater by cathodic stripping voltammetry with dihydroxynaphthalene. Anal. Chem. 2006, 78, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, N.; Xu, M.M.; Wang, J.; Jiang, C.; Song, G.; Wang, Y. Specific colorimetric detection of Fe3+ ions in aqueous solution by squaraine-based chemosensor. RSC Adv. 2018, 8, 34860–34866. [Google Scholar] [CrossRef] [Green Version]
- Adebayo, B.K.; Ayejuyo, S.; Okoro, H.K.; Ximba, B.J. Spectrophotometric determination of iron (III) in tap water using 8-hydoxyquinoline as a chromogenic reagent. Afr. J. Biotechnol. 2011, 10, 16051–16057. [Google Scholar] [CrossRef]
- Lashgari, N.; Badiei, A.; Mohammadi Ziarani, G. A Fluorescent Sensor for Al(III) and Colorimetric Sensor for Fe(III) and Fe(II) Based on a Novel 8-Hydroxyquinoline Derivative. J. Fluoresc. 2016, 26, 1885–1894. [Google Scholar] [CrossRef]
- Harathi, J.; Thenmozhi, K. AIE-active Schiff base compounds as fluorescent probes for the highly sensitive and selective detection of Fe3+ ions. Mater. Chem. Front. 2020, 4, 1471–1482. [Google Scholar] [CrossRef]
- Wei, D.; Sun, Y.; Yin, J.; Wei, G.; Du, Y. Design and application of Fe3+ probe for “naked-eye” colorimetric detection in fully aqueous system. Sens. Actuators B Chem. 2011, 160, 1316–1321. [Google Scholar] [CrossRef]
- Nizar, S.A.; Kobayashi, T.; Mohd Suah, F.B. An aminonaphthalene-based colorimetric and fluorescent sensor for selective recognition of Fe3+ in water. Luminescence 2020, 35, 1286–1295. [Google Scholar] [CrossRef]
- Jang, H.J.; Ahn, H.M.; Kim, M.S.; Kim, C. A highly selective colorimetric chemosensor for sequential detection of Fe3+ and pyrophosphate in aqueous solution. Tetrahedron 2017, 73, 6624–6631. [Google Scholar] [CrossRef]
- Wang, W.; Wei, J.; Liu, H.; Liu, Q.; Gao, Y. A novel colorimetric chemosensor based on quinoline for the sequential detection of Fe3+ and PPi in aqueous solution. Tetrahedron Lett. 2017, 58, 1025–1029. [Google Scholar] [CrossRef]
- Kao, M.H.; Wan, C.F.; Wu, A.T. A selective colorimetric chemosensor for Fe3+. Luminescence 2017, 32, 1561–1566. [Google Scholar] [CrossRef]
- Wang, L.; Li, W.; Zhi, W.; Huang, Y.; Han, J.; Wang, Y.; Ren, Y.; Ni, L. A new coumarin schiff based fluorescent-colorimetric chemosensor for dual monitoring of Zn2+ and Fe3+ in different solutions: An application to bio-imaging. Sens. Actuators B Chem. 2018, 260, 243–254. [Google Scholar] [CrossRef]
- Zhou, F.; Leng, T.H.; Liu, Y.J.; Wang, C.Y.; Shi, P.; Zhu, W.H. Water-soluble rhodamine-based chemosensor for Fe3+ with high sensitivity, selectivity and anti-interference capacity and its imaging application in living cells. Dye. Pigment. 2017, 142, 429–436. [Google Scholar] [CrossRef]
- Hao, E.; Meng, T.; Zhang, M.; Pang, W.; Zhou, Y.; Jiao, L. Solvent dependent fluorescent properties of a 1,2,3-triazole linked 8-hydroxyquinoline chemosensor: Tunable detection from zinc(II) to iron(III) in the CH3CN/H2O system. J. Phys. Chem. A 2011, 115, 8234–8241. [Google Scholar] [CrossRef]
- Liu, S.R.; Wu, S.P. New water-soluble highly selective fluorescent chemosensor for Fe (III) ions and its application to living cell imaging. Sens. Actuators B Chem. 2012, 171–172, 1110–1116. [Google Scholar] [CrossRef]
- Suryawanshi, S.B.; Mahajan, P.G.; Bodake, A.J.; Kolekar, G.B.; Patil, S.R. Carbazole based nanoprobe for selective recognition of Fe3 + ion in aqueous medium: Spectroscopic insight. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 183, 232–238. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, D.; Zhang, G.; Yang, J.; Dong, C.; Shuang, S. Glutathione capped silver nanoclusters-based fluorescent probe for highly sensitive detection of Fe3+. Sens. Actuators B Chem. 2014, 202, 631–637. [Google Scholar] [CrossRef]
- Vanjare, B.D.; Mahajan, P.G.; Hong, S.K.; Lee, K.H. Discriminating Chemosensor for Detection of Fe3+ in Aqueous Media by Fluorescence Quenching Methodology. Bull. Korean Chem. Soc. 2018, 39, 631–637. [Google Scholar] [CrossRef]
- Dong, L.; Wu, C.; Zeng, X.; Mu, L.; Xue, S.F.; Tao, Z.; Zhang, J.X. The synthesis of a rhodamine B schiff-base chemosensor and recognition properties for Fe3+ in neutral ethanol aqueous solution. Sens. Actuators B Chem. 2010, 145, 433–437. [Google Scholar] [CrossRef]
- Gupta, R.S.; Dudani, A.K. Species-specific differences in the toxicity of rhodamine 123 toward cultured mammalian cells. J. Cell. Physiol. 1987, 130, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, S.; Heinz, U.; Bartholomä, M.; Weyhermüller, T.; Nick, H.; Hegetschweiler, K. Complex Formation of ICL670 and Related Ligands with Fe III and Fe II. Eur. J. Inorg. Chem. 2004, 2004, 4177–4192. [Google Scholar] [CrossRef]
- Rouge, P.; Dassonville-Klimpt, A.; Cézard, C.; Boudesocque, S.; Ourouda, R.; Amant, C.; Gaboriau, F.; Forfar, I.; Guillon, J.; Guillon, E.; et al. Synthesis, physicochemical studies, molecular dynamics simulations, and metal-ion-dependent antiproliferative and antiangiogenic properties of cone ICL670-substituted calix[4]arenes. Chempluschem 2012, 77, 1001–1016. [Google Scholar] [CrossRef]
- Jo, T.G.; Jung, J.M.; Han, J.; Lim, M.H.; Kim, C. A single fluorescent chemosensor for multiple targets of Cu2+, Fe2+/3+ and Al3+ in living cells and a near-perfect aqueous solution. RSC Adv. 2017, 7, 28723–28732. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.B.; Zhang, P.; Wei, T.B.; Yao, H.; Lin, Q.; Zhang, Y.M. Highly selective fluorescent sensing for CN− in water: Utilization of the supramolecular self-assembly. Chem. Commun. 2013, 49, 7812–7814. [Google Scholar] [CrossRef]
- Simpson, P.V.; Schmidt, C.; Ott, I.; Bruhn, H.; Schatzschneider, U. Synthesis, Cellular Uptake and Biological Activity Against Pathogenic Microorganisms and Cancer Cells of Rhodium and Iridium N-Heterocyclic Carbene Complexes Bearing Charged Substituents. Eur. J. Inorg. Chem. 2013, 2013, 5547–5554. [Google Scholar] [CrossRef]
- Peng, Y.; Shi, Y.; Zhang, H.; Mine, Y.; Tsao, R. Anti-inflammatory and anti-oxidative activities of daidzein and its sulfonic acid ester derivatives. J. Funct. Foods 2017, 35, 635–640. [Google Scholar] [CrossRef]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Yan, K.-C.; Mangel, D.N.; Shang, Y.; Steinbrueck, A.; Han, H.-H.; Brewster, J.T.; Hu, X.-L.; Snelson, D.W.; Lynch, V.M.; et al. Deferasirox (ExJade): An FDA-Approved AIEgen Platform with Unique Photophysical Properties. J. Am. Chem. Soc. 2021, 143, 1278–1283. [Google Scholar] [CrossRef]
- Neurath, H.; Greenstein, J.P.; Putnam, F.W.; Erickson, J.O. The chemistry of protein denaturation. Chem. Rev. 1944, 34, 157–265. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hirose, M. Partially folded state of the disulfide-reduced form of human serum albumin as an intermediate for reversible denaturation. J. Biol. Chem. 1992, 267, 14753–14758. [Google Scholar] [CrossRef]
- Han, H.H.; Sedgwick, A.C.; Shang, Y.; Li, N.; Liu, T.; Li, B.H.; Yu, K.; Zang, Y.; Brewster, J.T.; Odyniec, M.L.; et al. Protein encapsulation: A new approach for improving the capability of small-molecule fluorogenic probes. Chem. Sci. 2020, 11, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Edwardson, T.G.W.; Tetter, S.; Hilvert, D. Two-tier supramolecular encapsulation of small molecules in a protein cage. Nat. Commun. 2020, 11, 5410. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Han, H.-H.; Sedgwick, A.C.; Williams, G.T.; Gwynne, L.; Brewster, J.T.; Bull, S.D.; Jenkins, A.T.A.; He, X.-P.; Schönherr, H.; et al. Protein Encapsulation: A Nanocarrier Approach to the Fluorescence Imaging of an Enzyme-Based Biomarker. Front. Chem. 2020, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Perrin, M.W.; Sedgwick, A.C.; Dunsky, E.Y.; Lynch, V.M.; He, X.P.; Sessler, J.L.; Arambula, J.F. Toward multifunctional anticancer therapeutics: Post-synthetic carbonate functionalisation of asymmetric Au(i) bis-N-heterocyclic carbenes. Chem. Commun. 2020, 56, 7877–7880. [Google Scholar] [CrossRef] [PubMed]
- Nusrat, S.; Siddiqi, M.K.; Zaman, M.; Zaidi, N.; Ajmal, M.R.; Alam, P.; Qadeer, A.; Abdelhameed, A.S.; Khan, R.H. A comprehensive spectroscopic and computational investigation to probe the interaction of antineoplastic drug nordihydroguaiaretic acid with serum albumins. PLoS ONE 2016, 11, e0158833. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of fluorescence spectroscopy Chapter 4: Time-Domain Lifetime Measurements. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006; pp. 97–156. ISBN 0387312781. [Google Scholar]
- Calvert, J.G.; Pitts, J.N. Photochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1966; pp. 626–627. ISBN 0471130907. [Google Scholar]
- Loza-Rosas, S.A.; Vázquez-Salgado, A.M.; Rivero, K.I.; Negrón, L.J.; Delgado, Y.; Benjamín-Rivera, J.A.; Vázquez-Maldonado, A.L.; Parks, T.B.; Munet-Colón, C.; Tinoco, A.D. Expanding the Therapeutic Potential of the Iron Chelator Deferasirox in the Development of Aqueous Stable Ti(IV) Anticancer Complexes. Inorg. Chem. 2017, 56, 7788–7802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure and Core-Scaffold | Sensing Method | Association Constant (Ka) | LOD for Fe3+ | Solvent System 1 | Ref. |
|---|---|---|---|---|---|
 based on deferasirox | Colorimetry and Fluorescence | 2.4 × 107 M−1 | 0.24 µM | 100% H2O | This work |
 based on squaraine | Colorimetry | 9.6 × 105 M−1 | 1 µM | 80% H2O 20% AcOH | [18] |
 based on coumarine | Colorimetry and Fluorescence | 9.4 × 103 M−1 | 5.38 µM | 80% EtOH 20% H2O | [27] |
 based on rhodamine | Colorimetry and Fluorescence | 4.7 × 108 M−1 | 0.28 µM | 50% H2O 50% DMSO | [28] |
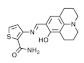 based on julolidine | Colorimetry | 9.9 × 104 M−1 | 0.51 µM | 100% H2O | [24] |
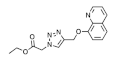 based on 8-hydroxyquinoline | Fluorescence | not reported | 1.23 µM | 95% MeCN 5% H2O | [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinbrueck, A.; Sedgwick, A.C.; Hwang, S.-M.; Sen, S.; Zhao, M.Y.; Huang, D.-Y.; Knoll, D.M.; Wang, Y.-Y.; Sessler, J.L. A Deferasirox Derivative That Acts as a Multifaceted Platform for the Detection and Quantification of Fe3+. Chemosensors 2021, 9, 68. https://doi.org/10.3390/chemosensors9040068
Steinbrueck A, Sedgwick AC, Hwang S-M, Sen S, Zhao MY, Huang D-Y, Knoll DM, Wang Y-Y, Sessler JL. A Deferasirox Derivative That Acts as a Multifaceted Platform for the Detection and Quantification of Fe3+. Chemosensors. 2021; 9(4):68. https://doi.org/10.3390/chemosensors9040068
Chicago/Turabian StyleSteinbrueck, Axel, Adam C. Sedgwick, Suh-Mi Hwang, Sajal Sen, Michael Y. Zhao, Dan-Ying Huang, Daniel M. Knoll, Yu-Ying Wang, and Jonathan L. Sessler. 2021. "A Deferasirox Derivative That Acts as a Multifaceted Platform for the Detection and Quantification of Fe3+" Chemosensors 9, no. 4: 68. https://doi.org/10.3390/chemosensors9040068
APA StyleSteinbrueck, A., Sedgwick, A. C., Hwang, S. -M., Sen, S., Zhao, M. Y., Huang, D. -Y., Knoll, D. M., Wang, Y. -Y., & Sessler, J. L. (2021). A Deferasirox Derivative That Acts as a Multifaceted Platform for the Detection and Quantification of Fe3+. Chemosensors, 9(4), 68. https://doi.org/10.3390/chemosensors9040068