
An Asymptomatic, Ectopic Mass as a Presentation of Adrenocortical Carcinoma Due to a Novel Germline TP53 p.Phe338Leu Tetramerisation Domain Variant
 , , , , , and
, , , , , and
Abstract
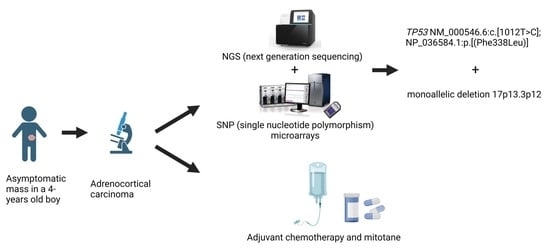
:
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient/Guardian Consent
2.2. Next-Generation Sequencing (NGS)
2.3. DNA Sequencing
2.4. Genomic Microarray Analysis
3. Case Description
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riedmeier, M.; Decarolis, B.; Haubitz, I.; Müller, S.; Uttinger, K.; Börner, K.; Reibetanz, J.; Wiegering, A.; Härtel, C.; Schlegel, P.G.; et al. Adrenocortical Carcinoma in Childhood: A Systematic Review. Cancers 2021, 13, 5266. [Google Scholar] [CrossRef] [PubMed]
- Virgone, C.; Roganovic, J.; Vorwerk, P.; Redlich, A.; Schneider, D.T.; Janic, D.; Bien, E.; López-Almaraz, R.; Godzinski, J.; Osterlundh, G.; et al. Adrenocortical tumours in children and adolescents: The EXPeRT/PARTNER diagnostic and therapeutic recommendations. Pediatr. Blood Cancer 2021, 68 (Suppl. 4), e29025. [Google Scholar] [CrossRef] [PubMed]
- Gulack, B.C.; Rialon, K.L.; Englum, B.R.; Kim, J.; Talbot, L.J.; Adibe, O.O.; Rice, H.E.; Tracy, E.T. Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB). J. Pediatr. Surg. 2016, 51, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Rivera, M.; Novotny, P.; Rodriguez, V.; Bancos, I.; Lteif, A. Adrenocortical Carcinoma in Children: A Clinicopathological Analysis of 41 Patients at the Mayo Clinic from 1950 to 2017. Horm. Res. Paediatr. 2018, 90, 8–18. [Google Scholar] [CrossRef]
- Pan, L.H.; Yen, C.C.; Huang, C.J.; Ng, X.N.; Lin, L.Y. Prognostic predictors of adrenocortical carcinoma: A single-center thirty-year experience. Front. Endocrinol. 2023, 14, 1134643. [Google Scholar] [CrossRef]
- Wang, R.; Solomon, B.; Luen, S.J.; Prall, O.W.J.; Khoo, C.; Gill, A.J.; Lewin, J.; Sachithanandan, N. Pitfalls and progress in adrenocortical carcinoma diagnosis: The utility of a multidisciplinary approach, immunohistochemistry and genomics. Endocrinol. Diabetes Metab. Case Rep. 2022, 2022, 21-0081. [Google Scholar] [CrossRef]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Kratz, C.P.; Freycon, C.; Maxwell, K.N.; Nichols, K.E.; Schiffman, J.D.; Evans, D.G.; Achatz, M.I.; Savage, S.A.; Weitzel, J.N.; Garber, J.E.; et al. Analysis of the Li-Fraumeni Spectrum Based on an International Germline TP53 Variant Data Set: An International Agency for Research on Cancer TP53 Database Analysis. JAMA Oncol. 2021, 7, 1800–1805. [Google Scholar] [CrossRef]
- Raymond, V.M.; Else, T.; Everett, J.N.; Long, J.M.; Gruber, S.B.; Hammer, G.D. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E119–E125. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: A children’s oncology group study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef]
- Challis, B.G.; Kandasamy, N.; Powlson, A.S.; Koulouri, O.; Annamalai, A.K.; Happerfield, L.; Marker, A.J.; Arends, M.J.; Nik-Zainal, S.; Gurnell, M. Familial Adrenocortical Carcinoma in Association with Lynch Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Grau, E.; Solanes, A.; Izquierdo, A.; Del Valle, J.; Carrato, C.; Pineda, M.; Dueñas, N.; Pujol, M.; Lázaro, C.; et al. Characteristics of Adrenocortical Carcinoma Associated with Lynch Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, N.Y.; Cho, W.K.; Yim, J.; Lee, J.W.; Kim, M.; Chung, J.H.; Jung, M.H.; Suh, B.K.; Ahn, M.B. Adrenocortical carcinoma and a sporadic MEN1 mutation in a 3-year-old girl: A case report. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Raymond, V.M.; Everett, J.N.; Furtado, L.V.; Gustafson, S.L.; Jungbluth, C.R.; Gruber, S.B.; Hammer, G.D.; Stoffel, E.M.; Greenson, J.K.; Giordano, T.J.; et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J. Clin. Oncol. 2013, 31, 3012–3018. [Google Scholar] [CrossRef]
- Torres, M.B.; Diggs, L.P.; Wei, J.S.; Khan, J.; Miettinen, M.; Fasaye, G.A.; Gillespie, A.; Widemann, B.C.; Kaplan, R.N.; Davis, J.L.; et al. Ataxia telangiectasia mutated germline pathogenic variant in adrenocortical carcinoma. Cancer Genet. 2021, 256–257, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.P.; Montgomery, K.W.; Tierney, J.; Gilbert, J.; Solórzano, C.C.; Idrees, K. Ectopic, retroperitoneal adrenocortical carcinoma in the setting of Lynch syndrome. Fam. Cancer 2018, 17, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, S.; Amano, R.; Onoda, N.; Noda, S.; Hirata, K.; Asano, Y.; Kurata, K.; Miura, K.; Yamazoe, S.; Kimura, K.; et al. Nonfunctional adrenocortical carcinoma initially presenting as retroperitoneal hemorrhage. BMC Surg. 2015, 15, 46. [Google Scholar] [CrossRef]
- Lenh, B.V.; Duc, N.M.; Tra My, T.T.; Minh, T.N.; Bang, L.V.; Linh, L.T.; Giang, B.V.; Thong, P.M. Non-functioning adrenocortical carcinoma. Radiol. Case Rep. 2021, 16, 1329–1334. [Google Scholar] [CrossRef]
- Lin, S.J.; Gao, Y.; Sun, C.J. Non-functional Adrenocortical Carcinoma in the Wall of the Small Bowel. Curr. Med. Imaging 2023. [Google Scholar] [CrossRef]
- Nie, L.; Wang, S.; Feng, Z.; Sun, Y.; Wang, Z.; Dang, Q.; Gao, A.; Lv, Y. Nonfunctional ectopic adrenocortical carcinoma in the lung: A case report and literature review. Front. Oncol. 2023, 13, 1100207. [Google Scholar] [CrossRef]
- Rashed, W.M.; Zekri, W.; Awad, M.; Taha, H.; Abdalla, B.; Alfaar, A.S. Nonfunctioning Adrenocortical Carcinoma in Pediatric Acute Lymphoblastic Leukemia: A Case Report of a Rare Multiple Primaries Combination. J. Pediatr. Hematol. Oncol. 2017, 39, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.H.; Chen, T.C.; Dai, S.H.; Zeng, Y.H. Case Report: Ectopic Adrenocortical Carcinoma in the Ovary. Front. Endocrinol. 2021, 12, 662377. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Scheithauer, B.W.; Erickson, L.A.; Jenkins, R.B.; Giannini, C. Ectopic low-grade adrenocortical carcinoma in the spinal region: Immunohistochemical and molecular cytogenetic study of a pediatric case. Am. J. Surg. Pathol. 2009, 33, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Riedmeier, M.; Thompson, L.D.R.; Molina, C.A.F.; Decarolis, B.; Härtel, C.; Schlegel, P.G.; Fassnacht, M.; Wiegering, V. Prognostic value of the Weiss and Wieneke (AFIP) scoring systems in pediatric ACC—A mini review. Endocr. Relat. Cancer 2023, 30. [Google Scholar] [CrossRef]
- Brenna, C.T.A.; Michaeli, O.; Wasserman, J.D.; Malkin, D. Clinical Outcomes of Children with Adrenocortical Carcinoma in the Context of Germline TP53 Status. J. Pediatr. Hematol. Oncol. 2021, 43, e635–e641. [Google Scholar] [CrossRef]
- Surakhy, M.; Wallace, M.; Bond, E.; Grochola, L.F.; Perez, H.; Di Giovannantonio, M.; Zhang, P.; Malkin, D.; Carter, H.; Parise, I.Z.S.; et al. A common polymorphism in the retinoic acid pathway modifies adrenocortical carcinoma age-dependent incidence. Br. J. Cancer 2020, 122, 1231–1241. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.D.; Gareth Evans, G.D. The European Reference Network GENTURIS. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Amadou, A.; Achatz, M.I.W.; Hainaut, P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: Temporal phases of Li-Fraumeni syndrome. Curr. Opin. Oncol. 2018, 30, 23–29. [Google Scholar] [CrossRef]
- Cornelius, A.; Foley, J.; Bond, J.; Nagulapally, A.B.; Steinbrecher, J.; Hendricks, W.P.D.; Rich, M.; Yendrembam, S.; Bergendahl, G.; Trent, J.M.; et al. Molecular Guided Therapy Provides Sustained Clinical Response in Refractory Choroid Plexus Carcinoma. Front. Pharmacol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walenciak, J.; Urbanska, Z.; Pastorczak, A.; Babol-Pokora, K.; Wypyszczak, K.; Bien, E.; Gawlowska-Marciniak, A.; Kobos, J.; Grajkowska, W.; Smyczynska, J.; et al. An Asymptomatic, Ectopic Mass as a Presentation of Adrenocortical Carcinoma Due to a Novel Germline TP53 p.Phe338Leu Tetramerisation Domain Variant. Children 2023, 10, 1793. https://doi.org/10.3390/children10111793
Walenciak J, Urbanska Z, Pastorczak A, Babol-Pokora K, Wypyszczak K, Bien E, Gawlowska-Marciniak A, Kobos J, Grajkowska W, Smyczynska J, et al. An Asymptomatic, Ectopic Mass as a Presentation of Adrenocortical Carcinoma Due to a Novel Germline TP53 p.Phe338Leu Tetramerisation Domain Variant. Children. 2023; 10(11):1793. https://doi.org/10.3390/children10111793
Chicago/Turabian StyleWalenciak, Justyna, Zuzanna Urbanska, Agata Pastorczak, Katarzyna Babol-Pokora, Kamila Wypyszczak, Ewa Bien, Aleksandra Gawlowska-Marciniak, Jozef Kobos, Wieslawa Grajkowska, Joanna Smyczynska, and et al. 2023. "An Asymptomatic, Ectopic Mass as a Presentation of Adrenocortical Carcinoma Due to a Novel Germline TP53 p.Phe338Leu Tetramerisation Domain Variant" Children 10, no. 11: 1793. https://doi.org/10.3390/children10111793
APA StyleWalenciak, J., Urbanska, Z., Pastorczak, A., Babol-Pokora, K., Wypyszczak, K., Bien, E., Gawlowska-Marciniak, A., Kobos, J., Grajkowska, W., Smyczynska, J., Mlynarski, W., & Janczar, S. (2023). An Asymptomatic, Ectopic Mass as a Presentation of Adrenocortical Carcinoma Due to a Novel Germline TP53 p.Phe338Leu Tetramerisation Domain Variant. Children, 10(11), 1793. https://doi.org/10.3390/children10111793