
Dental Phenotype with Minor Ectodermal Symptoms Suggestive of WNT10A Deficiency

 , , ,
, , ,
Abstract
:1. Introduction
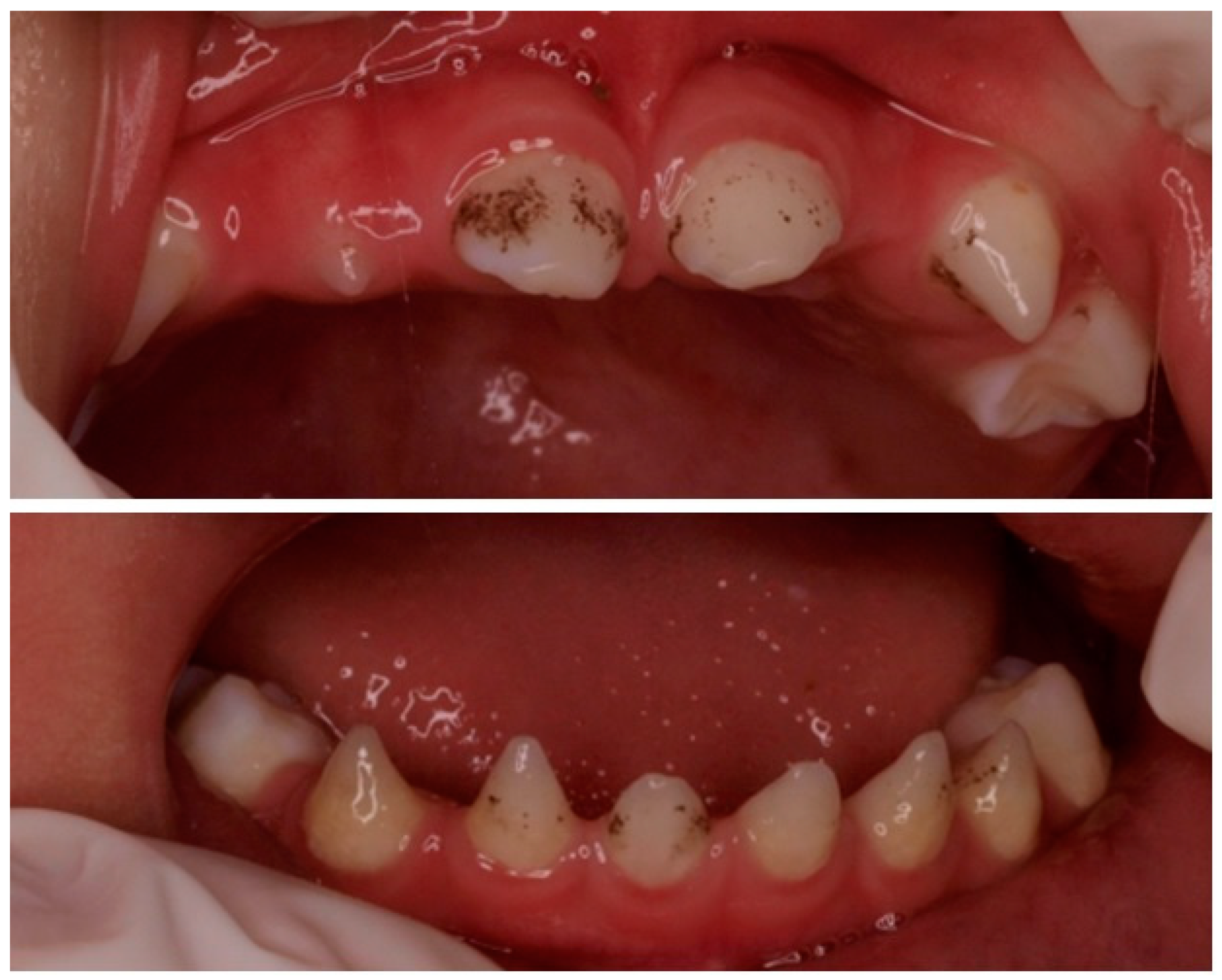
2. Clinical Case
2.1. Phenotype Study
2.2. Genetic Study
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patthey, C.; Gunhaga, L. Signaling pathways regulating ectodermal cell fate choices. Exp. Cell Res. 2014, 321, 11–16. [Google Scholar] [CrossRef]
- Naveau, A.; Seidel, K.; Klein, O.D. Tooth, hair and claw: Comparing epithelial stem cell niches of ectodermal appendages. Exp. Cell Res. 2014, 325, 96–103. [Google Scholar] [CrossRef]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasiasClassification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. A 2019, 179, 442–447. [Google Scholar] [CrossRef]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD and WNTA10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef]
- Itin, P.H. Etiology and pathogenesis of ectodermal dysplasias. Am. J. Med. Genet. A 2014, 164, 2472–2477. [Google Scholar] [CrossRef]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.-J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Chassaing, N.; Bourthoumieu, S.; Cossee, M.; Calvas, P.; Vincent, M.C. Mutations in EDAR account for one-quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum. Mutat. 2006, 27, 255–259. [Google Scholar] [CrossRef]
- Pinheiro, M.; Freire-Maia, N. Ectodermal dysplasias: A clinical classification and a causal review. Am. J. Med. Genet. A 1994, 53, 153–162. [Google Scholar] [CrossRef]
- Priolo, M.; Silengo, M.; Lerone, M.; Ravazzolo, R. Ectodermal dysplasias: Not only «skin» deep. Clin. Genet. 2000, 58, 415–430. [Google Scholar] [CrossRef]
- Lamartine, J. Towards a new classification of ectodermal dysplasia. Clin. Exp. Dermatol. 2003, 28, 351–355. [Google Scholar] [CrossRef]
- Irvine, A.D. Towards a unified classification of the ectodermal dysplasias: Opportunities outweigh challenges. Am. J. Med. Genet. A 2009, 149, 1970–1972. [Google Scholar] [CrossRef]
- Di Giovanna, J.J.; Priolo, M.; Itin, P. Approach towards a new classification for ectodermal dysplasias: Integration of the clinical and molecular knowledge. Am. J. Med. Genet. A 2009, 149, 2068–2070. [Google Scholar] [CrossRef]
- de Smalen, A.; van Nunen, D.P.F.; Hermus, R.R.; Ongkosuwito, E.M.; van Wijk, A.J.; Don Griot, J.P.W.; Breugem, C.C.; Kramer, G.J.C. Permanent tooth agenesis in non-syndromic Robin sequence and cleft palate: Prevalence and patterns. Clin. Oral Investig. 2017, 21, 2273–2281. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- rs3827760 (SNP)—Citations—Homo_Sapiens—Ensembl Genome Browser 108 [Internet]. Ensembl.org. Available online: https://oct2022.archive.ensembl.org/Homo_sapiens/Variation/Citations?db=core;r=2:108896645-108897645;v=rs3827760;vdb=variation;vf=183839455 (accessed on 25 October 2022).
- Fauzi, N.H.; Ardini, Y.D.; Zainuddin, Z.; Lestari, W. A review on non-syndromic tooth agenesis associated with PAX9 mutations. Jpn. Dent. Sci. Rev. 2018, 54, 30–36. [Google Scholar] [CrossRef]
- Doolan, B.J.; Onoufriadis, A.; Kantaputra, P.; McGrath, J.A. WNT I0 A, dermatology and dentistry. Brit. J. Dermatol. 2021, 185, 1105–1111. [Google Scholar] [CrossRef]
- Han, Y.; Wang, X.; Zheng, L.; Zhu, T.; Li, Y.; Hong, J.; Xu, C.; Wang, P.; Gao, M. Pathogenic EDA Mutations in Chinese Han Families With Hypohidrotic Ectodermal Dysplasia and Genotype-Phenotype: A Correlation Analysis. Front. Genet. 2020, 11, 21. [Google Scholar] [CrossRef]
- Okita, T.; Asano, N.; Yasuno, S.; Shimomura, Y. Functional studies for a dominant mutation in the EDAR gene responsible for hypohidrotic ectodermal dysplasia. J. Dermatol. 2019, 46, 710–715. [Google Scholar] [CrossRef]
- Chassaing, N.; Cluzeau, C.; Bal, E.; Guigue, P.; Vincent, M.C.; Viot, G.; Ginisty, D.; Munnich, A.A.; Calvas, P. Mutations in EDARADD account for a small proportion of hypohidrotic ectodermal dysplasia cases. Brit. J. Dermatol. 2010, 162, 1044–1048. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Khalifa, O.; Binamer, Y.M.; Almutawa, A.; Arold, S.T.; Zaidan, H.; Alkuraya, F.S. KDF1, encoding keratinocyte differentiation factor 1, is mutated in a multigenerational family with ectodermal dysplasia. Hum. Genet. 2017, 136, 99–105. [Google Scholar] [CrossRef]
- Manaspon, C.; Thaweesapphithak, S.; Osathanon, T.; Suphapeetiporn, K.; Porntaveetus, T.; Shotelersuk, V. A novel de novo mutation substantiates KDF1 as a gene causing ectodermal dysplasia. Br. J. Dermatol. 2019, 18, 419–420. [Google Scholar] [CrossRef]
- Yang, J.; Wang, S.K.; Choi, M.; Reid, B.M.; Hu, Y.; Lee, Y.L.; Herzog, C.R.; Kim-Berman, H.; Lee, M.; Benke, P.J.; et al. Taurodontism, variations in tooth number, and misshapened crowns in WNT10A null mice and human kindreds. Mol. Genet. Genomic. Med. 2015, 3, 40–58. [Google Scholar] [CrossRef]
- Martínez-Romero, M.C.; Ballesta-Martínez, M.J.; López-González, V.; GIEDE (Spanish multidisciplinary research group for ectodermal dysplasia). EDA, EDAR, EDARADD and WNT10A allelic variants in patients with ectodermal derivative impairment in the Spanish population. Orphanet. J. Rare Dis. 2019, 14, 281. [Google Scholar] [CrossRef]
- Tziotzios, C.; Petrof, G.; Liu, L.; Verma, A.; Wedgeworth, E.K.; Mellerio, J.E.; McGrath, J.A. Clinical features and WNT10A mutations in seven unrelated cases of Schopf-Schulz-Passarge syndrome. Br. J. Dermatol. 2014, 171, 1211–1214. [Google Scholar] [CrossRef]
- Riera-Monroig, J.; Martinez-Romero, M.C.; Alos, L.; Guillen-Navarro, E.; Mascaro, J.M., Jr. Eccrine Syringofibroadenoma as a clue for the diagnosis of Schopf-Schulz-Passarge syndrome in acquired palmoplantar keratoderma. J. Cutan. Pathol. 2020, 47, 987–989. [Google Scholar] [CrossRef]
- Mues, G.; Bonds, J.; Xiang, L.; Vieira, A.R.; Seymen, F.; Klein, O.; D’Souza, R.N. The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am. J. Med. Genet. A 2014, 164, 2455–2460. [Google Scholar] [CrossRef]
- Pagnan, N.A.; Visinoni, A.F. Update on ectodermal dysplasia clinical classification. Am. J. Med. Genet. A 2014, 164, 2415–2423. [Google Scholar] [CrossRef]
- Güven, Y.; Bal, E.; Altunoglu, U.; Yücel, E.; Hadj-Rabia, S.; Koruyucu, M.; Tuna, E.B.; Çıldır, S.; Aktören, O.; Bodemer, C.; et al. Turkish Ectodermal Dysplasia Cohort: From Phenotype to Genotype in 17 Families. Cytogenet. Genome Res. 2019, 157, 189–196. [Google Scholar] [CrossRef]
- Hsu, T.C.; Lee, J.Y.; Hsu, M.M.; Chao, S.C. Case report of Schopf-Schulz-Passarge syndrome resulting from a missense mutation, p.Arg104Cys, in WNT10A. J. Dermatol. 2018, 45, 475–478. [Google Scholar] [CrossRef]
- Zeng, B.; Zhao, Q.; Li, S.; Lu, H.; Lu, J.; Ma, L.; Zhao, W.; Yu, D. Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis. Genes 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wong, S.W.; Han, D.; Cai, T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019, 25, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.E.; Soufi, M.; Ruppert, V.; Schaefer, J.R.; von Domarus, H. Schopf-Schulz-Passarge Syndrome: Previously Unreported WNT10A Genotype and Phenotypes in 9 Family Members. Acta Derm.-Venereol. 2019, 99, 113–114. [Google Scholar] [CrossRef]
- Capalbo, A.; Alonso Valero, R.; Jimenez-Almazan, J.; Mir Pardo, P.; Fabiani, M.; Jiménez, D.; Simon, C.; Martin-Rodriguez, J. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLoS Genet. 2019, 15, e1008409. [Google Scholar] [CrossRef]
- Cluzeau, C.; Hadj-Rabia, S.; Bal, E.; Clauss, F.; Munnich, A.; Bodemer, C.; Headon, D.; Smahi, A. The EDAR370A allele attenuates the severity of hypohidrotic ectodermal dysplasia caused by EDA gene mutation. Br. J. Dermatol. 2012, 166, 678–681. [Google Scholar] [CrossRef]
- Chang, S.H.; Jobling, S.; Brennan, K.; Headon, D.J. Enhanced Edar signalling has pleiotropic effects on craniofacial and cutaneous glands. PLoS ONE 2009, 4, e7591. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPO_TERM_ID | HPO_TERM_NAME | Patient | Description |
|---|---|---|---|
| HP:0006344 | Abnormality of primary molar morphology | YES | Irregular coronal morphology and highly divergent roots. |
| HP:0006481 | Abnormality of primary teeth | YES | Front teeth are conical-shaped. |
| HP:0011056 | Agenesis of first permanent molar tooth | NO | |
| HP:0001798 | Anonychia | NO | |
| HP:0006482 | Abnormality of dental morphology | YES | |
| HP:0001792 | Small nails | NO | |
| HP:0002209 | Sparse scalp hair | NO | |
| HP:0000478 | Abnormality of the eye | NO | |
| HP:0010298 | Smooth tongue | YES | |
| HP:0011053 | Agenesis of mandibular premolar | YES | Also agenesis of the maxillary premolars. |
| HP:0008388 | Abnormal toenail morphology | NO | |
| HP:0000668 | Hypodontia | NO | |
| HP:0001806 | Onycholysis | NO | |
| HP:0031405 | Poroma | NO | |
| HP:0000975 | Hyperhidrosis | NO | |
| HP:0002671 | Basal cell carcinoma | NO | |
| HP:0008391 | Dystrophic fingernails | NO | |
| HP:0005216 | Impaired mastication | NO | |
| HP:0000007 | Autosomal recessive inheritance | ||
| HP:0000613 | Photophobia | NO | |
| HP:0001231 | Abnormal fingernail morphology | NO | |
| HP:0010764 | Short eyelashes | NO | |
| HP:0006342 | Peg-shaped maxillary lateral incisors | YES | |
| HP:0006289 | Agenesis of central incisor | NO | |
| HP:0009804 | Tooth agenesis | YES | 1.4,1.5, 2.2, 2.4, 2.5, 3.1, 3.2, 3.3, 3.4, 3.5, 4.1, 4.2, 4.3, 4.4, 4.5, 5.5, 6.2, 6.5,7.5, 8.5. |
| HP:0001596 | Alopecia | NO | |
| HP:0001810 | Dystrophic toenails | NO | |
| HP:0000951 | Abnormality of the skin | YES | Multiple hyperpigmented lentiginous macules <5 mm on the upper back and buttocks. Appearance was slightly atrophic with loss of fingerprints on the thumbs of the hands. |
| HP:0031454 | Apocrine hidrocystoma | NO | |
| HP:0000684 | Delayed eruption of teeth | NO | |
| HP:0000320 | Bird-like facies | NO | |
| HP:0002231 | Sparse body hair | NO | |
| HP:0011219 | Short face | NO | |
| HP:0000202 | Oral cleft | NO | |
| HP:0000685 | Hypoplasia of teeth | NO | |
| HP:0000958 | Dry skin | YES | |
| HP:0002860 | Squamous cell carcinoma | NO | |
| HP:0012472 | Eclabion | NO | |
| HP:0000968 | Ectodermal dysplasia | YES | |
| HP:0006297 | Enamel hypoplasia | NO | |
| HP:0002164 | Nail dysplasia | YES | Brittle toenails. |
| HP:0001595 | Abnormal hair morphology | NO | |
| HP:0011313 | Narrow nails | NO | |
| HP:0008070 | Sparse hair | NO | |
| HP:0001807 | Ridged nails | NO | |
| HP:0006323 | Premature loss of primary teeth | NO | |
| HP:0007380 | Facial telangiectasia | NO | |
| HP:0000677 | Oligodontia | YES | |
| HP:0000689 | Dental malocclusion | YES | |
| HP:0000691 | Microdontia | NO | |
| HP:0000696 | Delayed eruption of permanent teeth | NO | |
| HP:0100840 | Aplasia/hypoplasia of the eyebrow | NO | |
| HP:0002213 | Fine hair | NO | |
| HP:0007410 | Palmoplantar hyperhidrosis | NO | |
| HP:0010783 | Erythema | YES | Erythema and some fissures were observed in the balls of the feet, suggestive of atopic pulpitis. |
| HP:0007556 | Plantar hyperkeratosis | NO | |
| HP:0011359 | Dry hair | NO | |
| HP:0000679 | Taurodontia | NO | |
| HP:0000687 | Widely spaced teeth | YES | |
| HP:0000690 | Agenesis of maxillary lateral incisors | YES | Agenesis of a maxillary lateral incisor, temporary and permanent. |
| HP:0032152 | Keratosis pilaris | YES | Mild keratosis pilaris on the cheeks. |
| HP:0001816 | Thin nails | NO | |
| HP:0025114 | Hypergranulosis | NO | |
| HP:0000982 | Palmoplantar keratoderma | NO | |
| HP:0025092 | Epidermal acanthosis | NO | |
| HP:0006336 | Short dental root | NO | |
| HP:0100615 | Ovarian neoplasm | NO | |
| HP:0040162 | Orthokeratosis | NO | |
| HP:0045075 | Sparse eyebrow | YES | Sparse hair in eyebrow tail. |
| HP:0000966 | Hypohidrosis | NO | |
| HP:0006349 | Agenesis of permanent teeth | YES | |
| HP:0011051 | Agenesis of premolar | YES | |
| HP:0011078 | Abnormality of canine | YES |
| Cases Reported | Güven et al., 2019 [29] | Hsu et al., 2018 [30] | Yang et al., 2015 [34] | Yu et al., 2019 [32] | Zimmermann et al., 2017 [33] | Novel Case | |
|---|---|---|---|---|---|---|---|
| Sex | F | M | M | F | M | M | |
| Age at diagnosis (years) | DNA (child) | 54 | 8.5 | 14 | 53 | 11 | |
| Tooth agenesis | |||||||
| HP:0006482 | Abnormality of dental morphology | YES | YES | YES | YES | YES | YES |
| HP:0006349 | Agenesis of permanent teeth | YES | YES | YES | YES | YES | YES |
| HP:0000677 | Oligodontia | YES | YES | YES | YES | YES | YES |
| Sweating | |||||||
| HP:0000966 | Hipohidrosis | NO | NO | NO | YES | NO | NO |
| HP:0007410 | Palmoplantar hyperhidrosis | YES | NO | NO | NO | YES | NO |
| Skin | |||||||
| HP:0000958 | Dry skin | YES | YES | NO | YES | YES | YES |
| HP:0000982 | Palmoplantar keratoderma | NO | YES | NO | NO | YES | NO |
| HP:0031454 | Apocrine hidrocystoma | NO | YES | NO | NO | YES | NO |
| Hair | |||||||
| HP:0002209 | Sparse scalp hair | YES | YES | NO | NO | YES | NO |
| HP:0002231 | Sparse body hair | ND | YES | NO | NO | YES | YES |
| Nails | |||||||
| HP:0002164 | Nail dysplasia | YES | YES | NO | YES | YES | YES |
| Clinical Diagnosis | ED | SSPS | STHAG | OODD | SSPS | STHAG with mild ED | |
| Variants in WNT10A (NM_025216.3) | c. 310 C > A | c. 310 C > A | c. 310 C > A/c. 637 T > A | c. 742 C > T | c. 742 C > T/c. 321 C > A | c. 310 C > A/c. 742 C > T | |
| Protein change | p. (Arg104Cys) | p. (Arg104Cys) | p. (Arg104Cys)/p. (Gly213Ser) | p. (Arg248Ter) | p. (Arg248Ter)/p. (Cys107Ter) | p. (Arg104Cys)/p. (Arg248Ter) | |
| Zigosity | Homozygote | Homozygote | Compound heterozygous | Homozygote | Compound heterozygous | Compound heterozygous | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Martínez, V.-E.; Galiana-Vallés, X.; Zomeño-Alcalá, O.; Rodríguez-López, R.; Llena, C.; Martínez-Romero, M.d.C.; Guillén-Navarro, E. Dental Phenotype with Minor Ectodermal Symptoms Suggestive of WNT10A Deficiency. Children 2023, 10, 356. https://doi.org/10.3390/children10020356
García-Martínez V-E, Galiana-Vallés X, Zomeño-Alcalá O, Rodríguez-López R, Llena C, Martínez-Romero MdC, Guillén-Navarro E. Dental Phenotype with Minor Ectodermal Symptoms Suggestive of WNT10A Deficiency. Children. 2023; 10(2):356. https://doi.org/10.3390/children10020356
Chicago/Turabian StyleGarcía-Martínez, Victoria-Eugenia, Ximo Galiana-Vallés, Otilia Zomeño-Alcalá, Raquel Rodríguez-López, Carmen Llena, María del Carmen Martínez-Romero, and Encarna Guillén-Navarro. 2023. "Dental Phenotype with Minor Ectodermal Symptoms Suggestive of WNT10A Deficiency" Children 10, no. 2: 356. https://doi.org/10.3390/children10020356
APA StyleGarcía-Martínez, V.-E., Galiana-Vallés, X., Zomeño-Alcalá, O., Rodríguez-López, R., Llena, C., Martínez-Romero, M. d. C., & Guillén-Navarro, E. (2023). Dental Phenotype with Minor Ectodermal Symptoms Suggestive of WNT10A Deficiency. Children, 10(2), 356. https://doi.org/10.3390/children10020356