

Holoprosencephaly: Review of Embryology, Clinical Phenotypes, Etiology and Management

Abstract
:1. Introduction
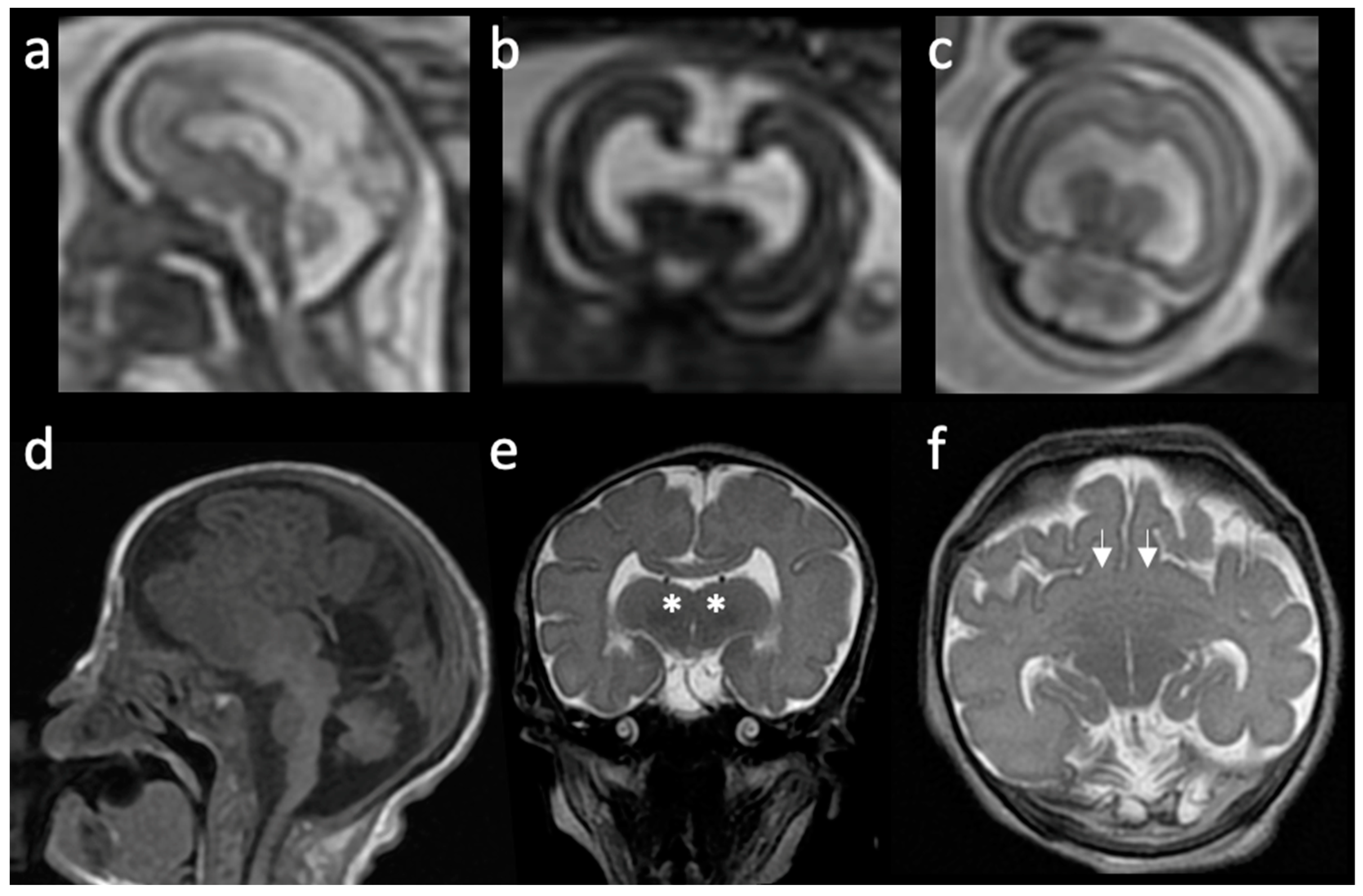
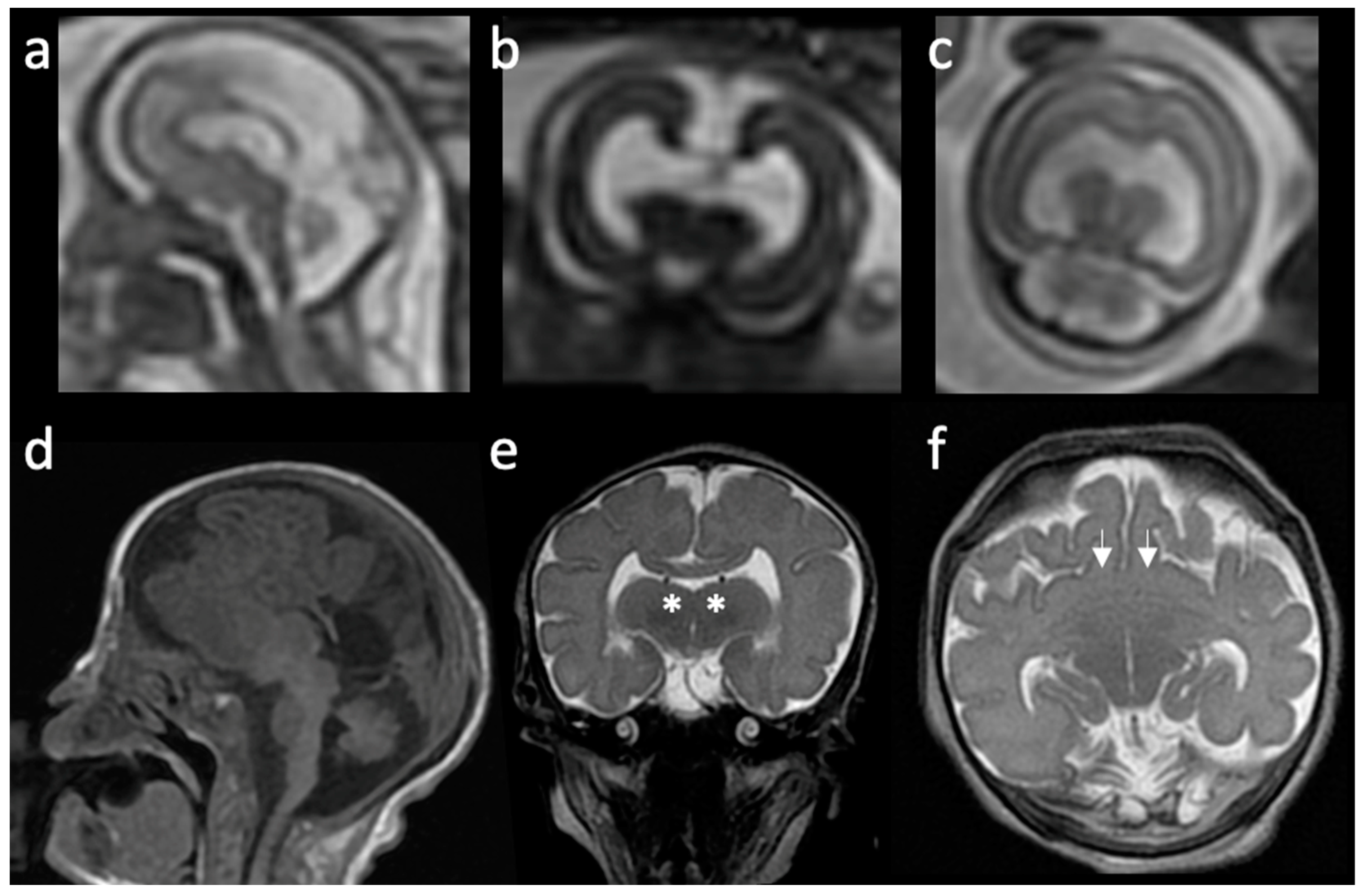
2. Overview of Prosencephalic Development
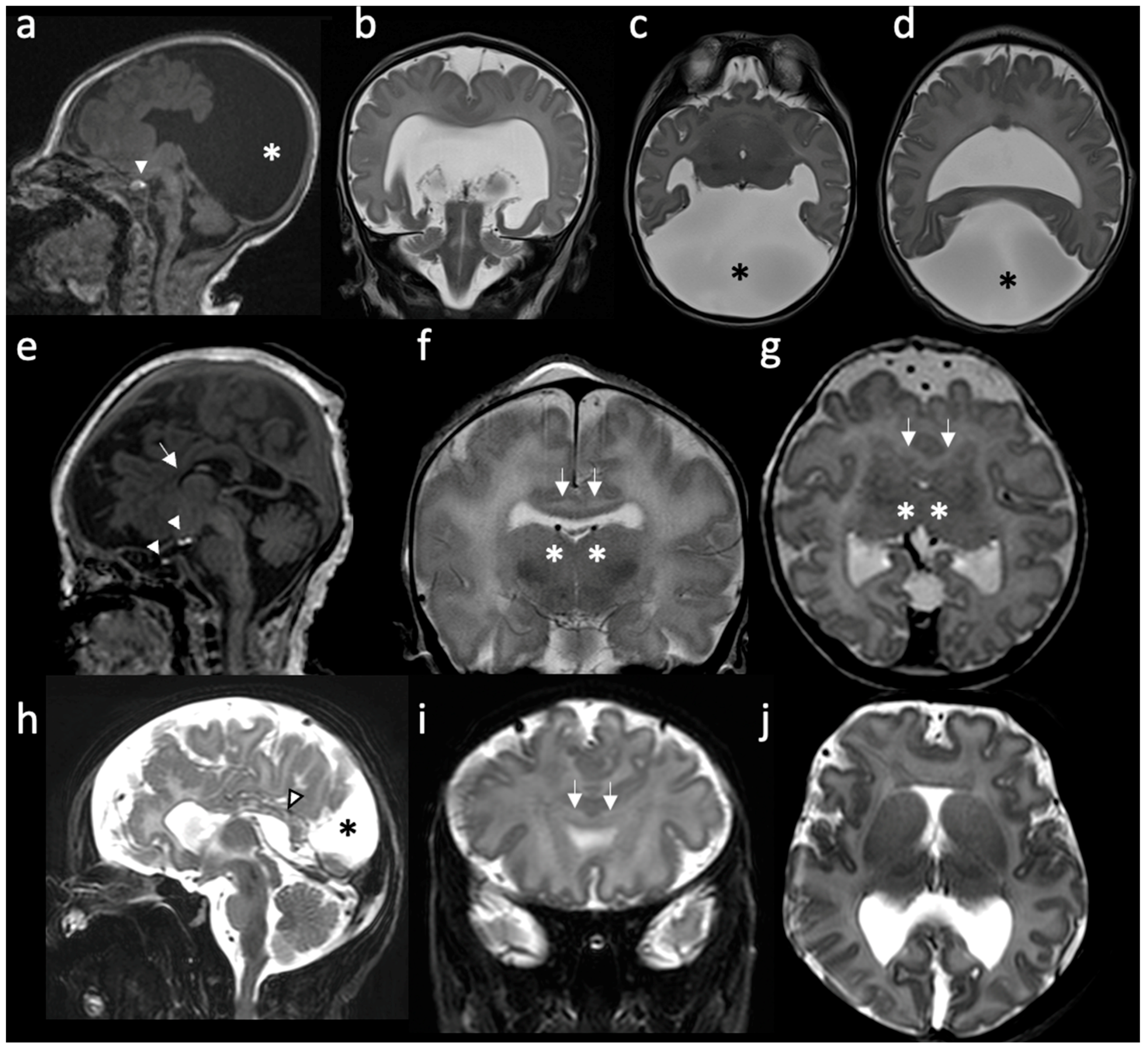
3. Radiologic Classification of HPE
4. Clinical Features
4.1. Craniofacial Defects and Developmental Outcomes
4.1.1. Alobar HPE
4.1.2. Semilobar HPE
4.1.3. Lobar HPE and MIH Variant
4.2. Survival
4.3. Other Common Clinical Features
5. Etiology
5.1. Genetic Causes
5.1.1. Chromosomal Anomalies
5.1.2. Monogenic Causes
- Non-syndromic monogenic causes
- Syndromic monogenic causes
- Smith–Lemli–Opitz syndrome (SLOS)
- Steinfield syndrome
- FGFR1-related syndromes
- Stromme syndrome
5.2. Non-Genetic Factors
6. Investigations and Management
6.1. Brain Imaging
6.2. Genetic Testing
6.3. Considerations Related to Prenatal Diagnosis and Genetic Counseling
6.4. Management
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monuki, E.S. The Morphogen Signaling Network in Forebrain Development and Holoprosencephaly. J. Neuropathol. Exp. Neurol. 2007, 66, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, S.; Gupta, V. Holoprosencephaly. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kruszka, P.; Gropman, A.L.; Muenke, M. Holoprosencephaly. In Cassidy and Allanson’s Management of Genetic Syndromes; Carey, J.C., Battaglia, A., Viskochil, D., Cassidy, S.B., Eds.; Wiley: New York, NY, USA, 2021; pp. 487–503. ISBN 978-1-119-43269-2. [Google Scholar]
- Dubourg, C.; Bendavid, C.; Pasquier, L.; Henry, C.; Odent, S.; David, V. Holoprosencephaly. Orphanet J. Rare Dis. 2007, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Grinblat, Y.; Lipinski, R.J. A Forebrain Undivided: Unleashing Model Organisms to Solve the Mysteries of Holoprosencephaly. Dev. Dyn. 2019, 248, 626–633. [Google Scholar] [CrossRef]
- Tekendo-Ngongang, C.; Muenke, M.; Kruszka, P. Holoprosencephaly Overview. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Raam, M.S.; Solomon, B.D.; Muenke, M. Holoprosencephaly: A Guide to Diagnosis and Clinical Management. Indian Pediatr. 2011, 48, 457–466. [Google Scholar] [CrossRef]
- Geng, X.; Oliver, G. Pathogenesis of Holoprosencephaly. J. Clin. Investig. 2009, 119, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, P.; Martinez, A.F.; Muenke, M. Molecular Testing in Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 187–193. [Google Scholar] [CrossRef]
- Volpe, P.; Campobasso, G.; De Robertis, V.; Rembouskos, G. Disorders of Prosencephalic Development. Prenat. Diagn. 2009, 29, 340–354. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Raybaud, C. Pediatric Neuroimaging; Wolters Kluwer Health: Philadelphia, PA, USA, 2012; ISBN 978-1-4511-5386-6. [Google Scholar]
- Andreu-Cervera, A.; Catala, M.; Schneider-Maunoury, S. Cilia, Ciliopathies and Hedgehog-Related Forebrain Developmental Disorders. Neurobiol. Dis. 2021, 150, 105236. [Google Scholar] [CrossRef]
- Ansari, A.; Bordoni, B. Embryology, Face. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Winter, T.C.; Kennedy, A.M.; Woodward, P.J. Holoprosencephaly: A Survey of the Entity, with Embryology and Fetal Imaging. Radiographics 2015, 35, 275–290. [Google Scholar] [CrossRef]
- Demyer, W.; Zeman, W.; Palmer, C.G. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly). Pediatrics 1964, 34, 256–263. [Google Scholar] [CrossRef]
- Leibovitz, Z.; Lerman-Sagie, T.; Haddad, L. Fetal Brain Development: Regulating Processes and Related Malformations. Life 2022, 12, 809. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Quint, D.J. Middle Interhemispheric Fusion: An Unusual Variant of Holoprosencephaly. Am. J. Neuroradiol. 1993, 14, 431–440. [Google Scholar]
- Hahn, J.S.; Barnes, P.D. Neuroimaging Advances in Holoprosencephaly: Refining the Spectrum of the Midline Malformation. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 120–132. [Google Scholar] [CrossRef]
- Marcorelles, P.; Laquerriere, A. Neuropathology of Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 109–119. [Google Scholar] [CrossRef]
- Schimmenti, L.A.; de la Cruz, J.; Lewis, R.A.; Karkera, J.D.; Manligas, G.S.; Roessler, E.; Muenke, M. Novel Mutation in Sonic Hedgehog in Non-Syndromic Colobomatous Microphthalmia. Am. J. Med. Genet. A 2003, 116A, 215–221. [Google Scholar] [CrossRef]
- Pilu, G.; Ambrosetto, P.; Sandri, F.; Tani, G.; Perolo, A.; Grisolia, G.; Ancora, G. Intraventricular Fused Fornices: A Specific Sign of Fetal Lobar Holoprosencephaly. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 1994, 4, 65–67. [Google Scholar] [CrossRef]
- Lewis, A.J.; Simon, E.M.; Barkovich, A.J.; Clegg, N.J.; Delgado, M.R.; Levey, E.; Hahn, J.S. Middle Interhemispheric Variant of Holoprosencephaly: A Distinct Cliniconeuroradiologic Subtype. Neurology 2002, 59, 1860–1865. [Google Scholar] [CrossRef]
- DeMyer W Holoprosencephaly. In Handbook of Clinical Neurology; North Holland: Amsterdam, The Netherlands, 1977; Volume 30, pp. 431–478.
- DeMyer W Holoprosencephaly (Cyclopia-Arhinencephaly). In Malformations; Elsevier: Amsterdam, The Netherlands; New York, NY, USA, 1987; Volume 50, pp. 225–244.
- Bernard, J.P.; Drummond, C.L.; Zaarour, P.; Molho, M.; Ville, Y. A New Clue to the Prenatal Diagnosis of Lobar Holoprosencephaly: The Abnormal Pathway of the Anterior Cerebral Artery Crawling under the Skull. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2002, 19, 605–607. [Google Scholar] [CrossRef]
- Levey, E.B.; Stashinko, E.; Clegg, N.J.; Delgado, M.R. Management of Children with Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 183–190. [Google Scholar] [CrossRef]
- Weiss, K.; Kruszka, P.; Guillen Sacoto, M.J.; Addissie, Y.A.; Hadley, D.W.; Hadsall, C.K.; Stokes, B.; Hu, P.; Roessler, E.; Solomon, B.; et al. In-Depth Investigations of Adolescents and Adults with Holoprosencephaly Identify Unique Characteristics. Genet. Med. 2018, 20, 14–23. [Google Scholar] [CrossRef]
- Lacbawan, F.; Solomon, B.D.; Roessler, E.; El-Jaick, K.; Domené, S.; Vélez, J.I.; Zhou, N.; Hadley, D.; Balog, J.Z.; Long, R.; et al. Clinical Spectrum of SIX3-Associated Mutations in Holoprosencephaly: Correlation between Genotype, Phenotype and Function. J. Med. Genet. 2009, 46, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Lacbawan, F.; Jain, M.; Domené, S.; Roessler, E.; Moore, C.; Dobyns, W.B.; Muenke, M. A Novel SIX3 Mutation Segregates with Holoprosencephaly in a Large Family. Am. J. Med. Genet. A 2009, 149A, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Kruszka, P.S.; Levey, E.; Muenke, M. Holoprosencephaly from Conception to Adulthood. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Bullen, P.J.; Rankin, J.M.; Robson, S.C. Investigation of the Epidemiology and Prenatal Diagnosis of Holoprosencephaly in the North of England. Am. J. Obstet. Gynecol. 2001, 184, 1256–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honey, E.M.; Bütow, K.W.; Zwahlen, R.A. Holoprosencephaly with Clefts: Data of 85 Patients, Treatment and Outcome: Part 1: History, Subdivisions, and Data on 85 Holoprosencephalic Cleft Patients. Ann. Maxillofac. Surg. 2019, 9, 140–145. [Google Scholar] [CrossRef]
- Croen, L.A.; Shaw, G.M.; Lammer, E.J. Holoprosencephaly: Epidemiologic and Clinical Characteristics of a California Population. Am. J. Med. Genet. 1996, 64, 465–472. [Google Scholar] [CrossRef]
- Barr, M.; Cohen, M.M. Holoprosencephaly Survival and Performance. Am. J. Med. Genet. 1999, 89, 116–120. [Google Scholar] [CrossRef]
- Pineda-Alvarez, D.E.; Dubourg, C.; David, V.; Roessler, E.; Muenke, M. Current Recommendations for the Molecular Evaluation of Newly Diagnosed Holoprosencephaly Patients. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Lacbawan, F.; Mercier, S.; Clegg, N.J.; Delgado, M.R.; Rosenbaum, K.; Dubourg, C.; David, V.; Olney, A.H.; Wehner, L.-E.; et al. Mutations in ZIC2 in Human Holoprosencephaly: Description of a Novel ZIC2 Specific Phenotype and Comprehensive Analysis of 157 Individuals. J. Med. Genet. 2010, 47, 513–524. [Google Scholar] [CrossRef]
- Hahn, J.S.; Hahn, S.M.; Kammann, H.; Barkovich, A.J.; Clegg, N.J.; Delgado, M.R.; Levey, E. Endocrine Disorders Associated with Holoprosencephaly. J. Pediatr. Endocrinol. Metab. JPEM 2005, 18, 935–941. [Google Scholar] [CrossRef]
- Roessler, E.; Vélez, J.I.; Zhou, N.; Muenke, M. Utilizing Prospective Sequence Analysis of SHH, ZIC2, SIX3 and TGIF in Holoprosencephaly Probands to Describe the Parameters Limiting the Observed Frequency of Mutant Gene×gene Interactions. Mol. Genet. Metab. 2012, 105, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Krauss, R.S. Modeling the Complex Etiology of Holoprosencephaly in Mice. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.S.; Hong, M. Gene-Environment Interactions and the Etiology of Birth Defects. Curr. Top. Dev. Biol. 2016, 116, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Tinker, S.C.; Gilboa, S.M.; Moore, C.A.; Waller, D.K.; Simeone, R.M.; Kim, S.Y.; Jamieson, D.J.; Botto, L.D.; Reefhuis, J. National Birth Defects Prevention Study Specific Birth Defects in Pregnancies of Women with Diabetes: National Birth Defects Prevention Study, 1997–2011. Am. J. Obstet. Gynecol. 2020, 222, 176.e1–176.e11. [Google Scholar] [CrossRef] [PubMed]
- Margaret Grace Norman. Congenital Malformations of the Brain: Pathologic, Embryologic, Clinical, Radiologic, and Genetic Aspects; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Addissie, Y.A.; Troia, A.; Wong, Z.C.; Everson, J.L.; Kozel, B.A.; Muenke, M.; Lipinski, R.J.; Malecki, K.M.C.; Kruszka, P. Identifying Environmental Risk Factors and gene–environment Interactions in Holoprosencephaly. Birth Defects Res. 2021, 113, 63–76. [Google Scholar] [CrossRef]
- Padmanabhan, R.; Shafiullah, M. Effect of Maternal Diabetes and Ethanol Interactions on Embryo Development in the Mouse. Mol. Cell Biochem. 2004, 261, 43–56. [Google Scholar] [CrossRef]
- Kauvar, E.F.; Muenke, M. Holoprosencephaly: Recommendations for Diagnosis and Management. Curr. Opin. Pediatr. 2010, 22, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, S.C.; Thakur, V.; Bronner-Fraser, M. Sonic Hedgehog Rescues Cranial Neural Crest from Cell Death Induced by Ethanol Exposure. Proc. Natl. Acad. Sci. USA 2002, 99, 10476–10481. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Krauss, R.S. Cdon Mutation and Fetal Ethanol Exposure Synergize to Produce Midline Signaling Defects and Holoprosencephaly Spectrum Disorders in Mice. PLoS Genet. 2012, 8, e1002999. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-X.; Yang, H.-T.; Zdanowicz, M.; Sicklick, J.K.; Qi, Y.; Camp, T.J.; Diehl, A.M. Fetal Alcohol Exposure Impairs Hedgehog Cholesterol Modification and Signaling. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, R.J.; Godin, E.A.; O’leary-Moore, S.K.; Parnell, S.E.; Sulik, K.K. Genesis of Teratogen-Induced Holoprosencephaly in Mice. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edison, R.; Muenke, M. The Interplay of Genetic and Environmental Factors in Craniofacial Morphogenesis: Holoprosencephaly and the Role of Cholesterol. Congenit. Anom. 2003, 43, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.A.; Rasmussen, S.A.; Siega-Riz, A.M.; Frías, J.L.; Honein, M.A. National Birth Defects Prevention Study Risk Factors for Non-Syndromic Holoprosencephaly in the National Birth Defects Prevention Study. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, C.; Kim, A.; Watrin, E.; de Tayrac, M.; Odent, S.; David, V.; Dupé, V. Recent Advances in Understanding Inheritance of Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.L.; Hughes, J.P.; Youngblood, L.G.; Sharpe-Stimac, M. Epidemiology of Holoprosencephaly and Phenotypic Characteristics of Affected Children: New York State, 1984-1989. Am. J. Med. Genet. 1997, 73, 217–226. [Google Scholar] [CrossRef]
- Hu, T.; Kruszka, P.; Martinez, A.F.; Ming, J.E.; Shabason, E.K.; Raam, M.S.; Shaikh, T.H.; Pineda-Alvarez, D.E.; Muenke, M. Cytogenetics and Holoprosencephaly: A Chromosomal Microarray Study of 222 Individuals with Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 175–186. [Google Scholar] [CrossRef]
- Kruszka, P.; Muenke, M. Syndromes Associated with Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 229–237. [Google Scholar] [CrossRef]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog Receptor Function during Craniofacial Development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef] [Green Version]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the Human Sonic Hedgehog Gene Cause Holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef]
- Roessler, E.; Muenke, M. Holoprosencephaly: A Paradigm for the Complex Genetics of Brain Development. J. Inherit. Metab. Dis. 1998, 21, 481–497. [Google Scholar] [CrossRef]
- Cohen, M.M. The Hedgehog Signaling Network. Am. J. Med. Genet. Part A 2003, 123A, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Bear, K.A.; Wyllie, A.; Keaton, A.A.; Dubourg, C.; David, V.; Mercier, S.; Odent, S.; Hehr, U.; Paulussen, A.; et al. Genotypic and Phenotypic Analysis of 396 Individuals with Mutations in Sonic Hedgehog. J. Med. Genet. 2012, 49, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.; Leskow, F.C.; El-Jaick, K.; Roessler, E.; Muenke, M.; Yocum, A.; Dubourg, C.; Li, X.; Geng, X.; Oliver, G.; et al. Regulation of a Remote Shh Forebrain Enhancer by the Six3 Homeoprotein. Nat. Genet. 2008, 40, 1348–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.Y.; Odent, S.; David, V.; Blayau, M.; Dubourg, C.; Apacik, C.; Delgado, M.A.; Hall, B.D.; Reynolds, J.F.; Sommer, A.; et al. Holoprosencephaly Due to Mutations in ZIC2: Alanine Tract Expansion Mutations May Be Caused by Parental Somatic Recombination. Hum. Mol. Genet. 2001, 10, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Du, Y.-Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-Function Mutations in the Human GLI2 Gene Are Associated with Pituitary Anomalies and Holoprosencephaly-like Features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [Green Version]
- Rahimov, F.; Ribeiro, L.A.; de Miranda, E.; Richieri-Costa, A.; Murray, J.C. GLI2 Mutations in Four Brazilian Patients: How Wide Is the Phenotypic Spectrum? Am. J. Med. Genet. Part A 2006, 140, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Bertolacini, C.D.P.; Ribeiro-Bicudo, L.A.; Petrin, A.; Richieri-Costa, A.; Murray, J.C. Clinical Findings in Patients with GLI2 Mutations--Phenotypic Variability. Clin. Genet. 2012, 81, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.M. Perspectives on Holoprosencephaly: Part I. Epidemiology, Genetics, and Syndromology. Teratology 1989, 40, 211–235. [Google Scholar] [CrossRef]
- Weaver, D.D.; Solomon, B.D.; Akin-Samson, K.; Kelley, R.I.; Muenke, M. Cyclopia (Synophthalmia) in Smith-Lemli-Opitz Syndrome: First Reported Case and Consideration of Mechanism. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 142–145. [Google Scholar] [CrossRef] [Green Version]
- Caruso, P.A.; Poussaint, T.Y.; Tzika, A.A.; Zurakowski, D.; Astrakas, L.G.; Elias, E.R.; Bay, C.; Irons, M.B. MRI and 1H MRS Findings in Smith-Lemli-Opitz Syndrome. Neuroradiology 2004, 46, 3–14. [Google Scholar] [CrossRef]
- Kelley, R.L.; Roessler, E.; Hennekam, R.C.; Feldman, G.L.; Kosaki, K.; Jones, M.C.; Palumbos, J.C.; Muenke, M. Holoprosencephaly in RSH/Smith-Lemli-Opitz Syndrome: Does Abnormal Cholesterol Metabolism Affect the Function of Sonic Hedgehog? Am. J. Med. Genet. 1996, 66, 478–484. [Google Scholar] [CrossRef]
- Nowaczyk, M.J.; Farrell, S.A.; Sirkin, W.L.; Velsher, L.; Krakowiak, P.A.; Waye, J.S.; Porter, F.D. Smith-Lemli-Opitz (RHS) Syndrome: Holoprosencephaly and Homozygous IVS8-1G-->C Genotype. Am. J. Med. Genet. 2001, 103, 75–80. [Google Scholar] [CrossRef]
- Travessa, A.; Dias, P.; Rocha, P.; Sousa, A.B. Prenatal Diagnosis of Holoprosencephaly Associated with Smith-Lemli-Opitz Syndrome (SLOS) in a 46,XX Fetus. Taiwan. J. Obstet. Gynecol. 2017, 56, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Muenke, M. Abnormal Sterol Metabolism in Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.E.; Robertson, L.; Maniyar, A.; Shammas, C.; Phelan, M.M.; Vasudevan, P.C.; Tanteles, G.A. Microform Holoprosencephaly with Bilateral Congenital Elbow Dislocation; Increasing the Phenotypic Spectrum of Steinfeld Syndrome. Am. J. Med. Genet. Part A 2016, 170, 754–759. [Google Scholar] [CrossRef]
- Dubourg, C.; Carré, W.; Hamdi-Rozé, H.; Mouden, C.; Roume, J.; Abdelmajid, B.; Amram, D.; Baumann, C.; Chassaing, N.; Coubes, C.; et al. Mutational Spectrum in Holoprosencephaly Shows That FGF Is a New Major Signaling Pathway. Hum. Mutat. 2016, 37, 1329–1339. [Google Scholar] [CrossRef]
- Simonis, N.; Migeotte, I.; Lambert, N.; Perazzolo, C.; de Silva, D.C.; Dimitrov, B.; Heinrichs, C.; Janssens, S.; Kerr, B.; Mortier, G.; et al. FGFR1 Mutations Cause Hartsfield Syndrome, the Unique Association of Holoprosencephaly and Ectrodactyly. J. Med. Genet. 2013, 50, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Hu, P.; Marino, J.; Hufnagel, S.B.; Hopkin, R.J.; Toromanović, A.; Richieri-Costa, A.; Ribeiro-Bicudo, L.A.; Kruszka, P.; Roessler, E.; et al. Dominant-Negative Kinase Domain Mutations in FGFR1 Can Explain the Clinical Severity of Hartsfield Syndrome. Hum. Mol. Genet. 2016, 25, 1912–1922. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, P.; Petracca, A.; Maggi, R.; Biagini, T.; Nardella, G.; Sacco, M.C.; Di Schiavi, E.; Carella, M.; Micale, L.; Castori, M. A Novel Dominant-Negative FGFR1 Variant Causes Hartsfield Syndrome by Deregulating RAS/ERK1/2 Pathway. Eur. J. Hum. Genet. 2019, 27, 1113–1120. [Google Scholar] [CrossRef]
- Alghamdi, M.; Alkhamis, W.H.; Bashiri, F.A.; Jamjoom, D.; Al-Nafisah, G.; Tahir, A.; Abdouelhoda, M. Expanding the Phenotype and the Genotype of Stromme Syndrome: A Novel Variant of the CENPF Gene and Literature Review. Eur. J. Med. Genet. 2020, 63, 103844. [Google Scholar] [CrossRef]
- Waters, A.M.; Asfahani, R.; Carroll, P.; Bicknell, L.; Lescai, F.; Bright, A.; Chanudet, E.; Brooks, A.; Christou-Savina, S.; Osman, G.; et al. The Kinetochore Protein, CENPF, Is Mutated in Human Ciliopathy and Microcephaly Phenotypes. J. Med. Genet. 2015, 52, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.Y.; Rasmussen, S.A. Non-Genetic Risk Factors for Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 73–85. [Google Scholar] [CrossRef]
- Croen, L.A.; Shaw, G.M.; Lammer, E.J. Risk Factors for Cytogenetically Normal Holoprosencephaly in California: A Popula-tion-Based Case-Control Study. Am. J. Med. Genet. 2000, 90, 320–325. [Google Scholar] [CrossRef]
- Kayembe-Kitenge, T.; Kasole Lubala, T.; Musa Obadia, P.; Katoto Chimusa, P.; Katshiez Nawej, C.; Banza Lubaba Nkulu, C.; Devriendt, K.; Nemery, B. Holoprosencephaly: A Case Series from an Area with High Mining-Related Pollution. Birth Defects Res. 2019, 111, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Kousa, Y.A.; du Plessis, A.J.; Vezina, G. Prenatal Diagnosis of Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.; Hui, L. First and Second Trimester Screening for Fetal Structural Anomalies. Semin. Fetal. Neonatal Med. 2018, 23, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Jarvis, D. In Utero MR Imaging of Fetal Holoprosencephaly: A Structured Approach to Diagnosis and Classification. Am. J. Neuroradiol. 2016, 37, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Balog, J.Z.; Hadley, D.; Gropman, A.L.; Nandagopal, R.; Han, J.C.; Hahn, J.S.; Blain, D.; Brooks, B.; et al. Compound Heterozygosity for Mutations in PAX6 in a Patient with Complex Brain Anomaly, Neonatal Diabetes Mellitus, and Microophthalmia. Am. J. Med. Genet. Part A 2009, 149A, 2543–2546. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Rosenbaum, K.N.; Meck, J.M.; Muenke, M. Holoprosencephaly Due to Numeric Chromosome Abnormalities. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 146–148. [Google Scholar] [CrossRef]
- Solomon, B.D.; Retterer, K.; Juusola, J. Holoprosencephaly: A Clinical Genomics Perspective. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Hadley, D.W.; Kruszka, P.; Muenke, M. Challenging Issues Arising in Counseling Families Experiencing Holoprosencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Elfarawi, H.; Tolusso, L.; McGowan, M.L.; Cortezzo, D.; Vawter-Lee, M. Alobar Holoprosencephaly: Exploring Mothers’ Perspectives on Prenatal Decision-Making and Prognostication. Prenat. Diagn. 2022, 42, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Redlinger-Grosse, K.; Bernhardt, B.A.; Berg, K.; Muenke, M.; Biesecker, B.B. The Decision to Continue: The Experiences and Needs of Parents Who Receive a Prenatal Diagnosis of Holoprosencephaly. Am. J. Med. Genet. 2002, 112, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Pond, E.; Dimond, R. Reproductive Decision Making: Interviews with Mothers of Children with Undiagnosed Developmental Delay. J. Community Genet. 2018, 9, 315–325. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Type | Main Features | References |
|---|---|---|
| Alobar | Absent separation of the cerebral hemispheres; Single “monoventricle”; Agenesis of the corpus callosum, absent third ventricle; Fusion of thalami and basal ganglia; Dorsal cyst is frequent; Significant midline facial defects. | [6,14,16,19,20] |
| Semilobar | Anterior lobes fail to separate; Interhemispheric fissure detected only posteriorly; Small, partially-formed third ventricle is often noted; Dorsal cyst may also be present; Midline craniofacial defects may be present or only subtle facial abnormalities. | [6,16] |
| Lobar | Only the most rostral-inferior parts of the frontal lobes are fused; Septum pellucidum is usually absent; Posterior half of the corpus callosum is formed; Varying degrees of basal ganglia and thalamic fusion; Midline craniofacial defects often absent or mild. | [6,16,21] |
| Middle interhemispheric variant (syntelencephaly) | Failure of separation of the posterior frontal and parietal lobes; Variable lack of cleavage of the basal ganglia and thalami; Absence of the body but presence of the genu and splenium of the corpus callosum. | [6,16,17,22] |
| Septopreoptic (minimal form) | Midline fusion restricted to the septal region or preoptic region of the telencephalon. | [16] |
| Microform | Only HPE-related subtle craniofacial anomalies; No structural brain defects on imaging. | [16] |
| Genetic Causes | |||
|---|---|---|---|
| Syndromic | Non-Syndromic | ||
| Chromosomal | Monogenic | ||
| Aneuploidies | Structural Abnormalities | ||
| Trisomy 13 (Patau syndrome) Trisomy 18 (Edwards syndrome) | del or dup(13q) del(18p) del(7)(q36) dup(3)(p24-pter) del(2)(p21) del(21)(q22.3) | CDON-Steinfeld syndrome | SHH (AD, MIM#142945) |
| (AD, MIM#184705) | ZIC2 (AD, MIM#609637) | ||
| FGFR1-Kallmann syndrome 2 | SIX3 (AD, MIM#157170) | ||
| (AD, MIM# #147950) | TGIF1 (AD, MIM#142946) | ||
| FGFR1-Hartsfield syndrome | GLI2 (AD, MIM#610829) | ||
| (AD, MIM#615465) | FGF8 (AD, MIM#612702) | ||
| CENPF-Stromme syndrome | FGFR1 (AD, MIM#147950) | ||
| (AR, MIM#243605) | DISP1 (AD, MIM#612530) | ||
| DHCR7-Smith-Lemli-Optiz syndrome | DLL1 (AD, MIM#618709) | ||
| (AR, MIM#270400) | CDON (AR, MIM#614226) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malta, M.; AlMutiri, R.; Martin, C.S.; Srour, M. Holoprosencephaly: Review of Embryology, Clinical Phenotypes, Etiology and Management. Children 2023, 10, 647. https://doi.org/10.3390/children10040647
Malta M, AlMutiri R, Martin CS, Srour M. Holoprosencephaly: Review of Embryology, Clinical Phenotypes, Etiology and Management. Children. 2023; 10(4):647. https://doi.org/10.3390/children10040647
Chicago/Turabian StyleMalta, Maísa, Rowim AlMutiri, Christine Saint Martin, and Myriam Srour. 2023. "Holoprosencephaly: Review of Embryology, Clinical Phenotypes, Etiology and Management" Children 10, no. 4: 647. https://doi.org/10.3390/children10040647
APA StyleMalta, M., AlMutiri, R., Martin, C. S., & Srour, M. (2023). Holoprosencephaly: Review of Embryology, Clinical Phenotypes, Etiology and Management. Children, 10(4), 647. https://doi.org/10.3390/children10040647