
Interstitial Lung Disease in Neonates: A Long Road Is Being Paved



Abstract
:1. Introduction
2. Relevant Disorders in Infants Aged from 0 to 2 Years Old
2.1. Diffuse Developmental Disorders
2.2. Alveolar Growth Disorders
2.3. Surfactant Disorders
2.4. Pulmonary Interstitial Glycogenosis (PIG)
2.5. Neuroendocrine Cell Hyperplasia of Infancy (NEHI)
3. Rare Disorders in Infants Aged from 0 to 2 Years Old
3.1. Idiopathic Fibrosing Alveolitis
3.2. Desquamative Interstitial Pneumonitis (DIP)
4. Cases Presentation
4.1. Case 1:
4.2. Case 2:
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spagnolo, P.; Bush, A. Interstitial Lung Disease in Children Younger Than 2 Years. Pediatrics 2016, 137, e20152725. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, G.H.; Young, L.R.; Deterding, R.R.; Fan, L.L.; Dell, S.D.; Bean, J.A.; Brody, A.S.; Nogee, L.M.; Trapnell, B.C.; Langston, C.; et al. Diffuse lung disease in young children: Application of a novel classification scheme. Am. J. Respir. Crit. Care Med. 2007, 176, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Nogee, L.M. Interstitial lung disease in newborns. Semin. Fetal Neonatal Med. 2017, 22, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Langston, C.; Fan, L.L. Interstitial lung disease in children. Curr. Opin. Pediatr. 2011, 23, 325–331. [Google Scholar] [CrossRef]
- Clement, A.; Eber, E. Interstitial lung diseases in infants and children. Eur. Respir. J. 2008, 31, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, F.; Maiese, A.; Spina, F.; Visi, G.; La Russa, R.; Santoro, P.; Pignotti, M.S.; Frati, P.; Fineschi, V. Idiopathic Pulmonary Hemorrhage in Infancy: A Case Report and Literature Review. Diagnostics 2023, 13, 1270. [Google Scholar] [CrossRef]
- Clement, A. Task force on chronic interstitial lung disease in immunocompetent children. Eur. Respir. J. 2004, 24, 686–697. [Google Scholar] [CrossRef]
- Fan, L.L.; Langston, C. Pediatric interstitial lung disease: Children are not small adults. Am. J. Respir. Crit. Care Med. 2002, 165, 1466–1467. [Google Scholar] [CrossRef]
- Glasser, S.W.; Hardie, W.D.; Hagood, J.S. Pathogenesis of Interstitial Lung Disease in Children and Adults. Pediatr. Allergy Immunol. Pulmonol. 2010, 23, 9–14. [Google Scholar] [CrossRef]
- Deterding, R. Evaluating infants and children with interstitial lung disease. Semin. Respir. Crit. Care Med. 2007, 28, 333–341. [Google Scholar] [CrossRef]
- Rice, A.; Tran-Dang, M.A.; Bush, A.; Nicholson, A.G. Diffuse lung disease in infancy and childhood: Expanding the chILD classifi cation. Histopathology 2013, 63, 743–755. [Google Scholar] [CrossRef]
- Langston, C.; Dishop, M.K. Diffuse lung disease in infancy: A proposed classification applied to 259 diagnostic biopsies. Pediatr. Dev. Pathol. 2009, 12, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y. Interstitial lung disease in infants: New classification system, imaging technique, clinical presentation and imaging findings. Pediatr. Radiol. 2013, 43, 3–13, quiz p. 128–129. [Google Scholar] [CrossRef] [PubMed]
- Guillerman, R.P.; Brody, A.S. Contemporary perspectives on pediatric diffuse lung disease. Radiol. Clin. N. Am. 2011, 49, 847–868. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.A.; Shannon, D.C.; Mark, E.J. Cellular interstitial pneumonitis in infants. A clinicopathologic study. Chest 1992, 101, 1065–1069. [Google Scholar] [CrossRef]
- Canakis, A.M.; Cutz, E.; Manson, D.; O’Brodovich, H. Pulmonary interstitial glycogenosis: A new variant of neonatal interstitial lung disease. Am. J. Respir. Crit. Care Med. 2002, 165, 1557–1565. [Google Scholar] [CrossRef]
- Lanfranchi, M.; Allbery, S.M.; Wheelock, L.; Perry, D. Pulmonary interstitial glycogenosis. Pediatr. Radiol. 2010, 40, 361–365. [Google Scholar] [CrossRef]
- Castillo, M.; Vade, A.; Lim-Dunham, J.E.; Masuda, E.; Massarani-Wafai, R. Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: Radiographic and pathologic correlation. Pediatr. Radiol. 2010, 40, 1562–1565. [Google Scholar] [CrossRef]
- Deterding, R.R.; Pye, C.; Fan, L.L.; Langston, C. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr. Pulmonol. 2005, 40, 157–165. [Google Scholar] [CrossRef]
- Guillerman, R.P. Imaging of childhood interstitial lung disease. Pediatr. Allergy Immunol. Pulmonol. 2010, 23, 43–68. [Google Scholar] [CrossRef]
- Young, L.R.; Deutsch, G.H.; Bokulic, R.E.; Brody, A.S.; Nogee, L.M. A mutation in TTF1/NKX2.1 is associated with familial neuroendocrine cell hyperplasia of infancy. Chest 2013, 144, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Lukkarinen, H.; Pelkonen, A.; Lohi, J.; Malmström, K.; Malmberg, L.P.; Kajosaari, M.; Lindahl, H.; Föhr, A.; Ruuskanen, O.; Mäkelä, M. Neuroendocrine cell hyperplasia of infancy: A prospective follow-up of nine children. Arch. Dis. Child. 2013, 98, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Deterding, R.R.; Langston, C. Pediatric interstitial lung disease revisited. Pediatr. Pulmonol. 2004, 38, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Osika, E.; Muller, M.H.; Boccon-Gibod, L.; Fauroux, B.; Sardet, A.; Grosskopf, C.; Couvreur, J.; Tournier, G.; Clement, A. Idiopathic pulmonary fibrosis in infants. Pediatr. Pulmonol. 1997, 23, 49–54. [Google Scholar] [CrossRef]
- Fan, L.L.; Kozinetz, C.A. Factors influencing survival in children with chronic interstitial lung disease. Am. J. Respir. Crit. Care Med. 1997, 156, 939–942. [Google Scholar] [CrossRef]
- Noble, P.W. Idiopathic pulmonary fibrosis: Natural history and prognosis. Clin. Chest Med. 2006, 27, S11–S16. [Google Scholar] [CrossRef]
- Stillwell, P.C.; Norris, D.G.; O’Connell, E.J.; Rosenow, E.C., III; Weiland, L.H.; Harrison, E.G., Jr. Desquamative interstitial pneumonitis in children. Chest 1980, 77, 165–171. [Google Scholar] [CrossRef]
- Carrington, C.B.; Gaensler, E.A.; Coutu, R.E.; FitzGerald, M.X.; Gupta, R.G. Natural history and treated course of usual and desquamative interstitial pneumonia. N. Engl. J. Med. 1978, 298, 801–809. [Google Scholar] [CrossRef]
- Hewitt, C.J.; Hull, D.; Keeling, J.W. Fibrosing alveolitis in infancy and childhood. Arch. Dis. Child. 1977, 52, 22–37. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Day of Life (DOL) | ||||||
|---|---|---|---|---|---|---|
| DOL 1 | DOL 2 | DOL 5 | DOL 10 | DOL13 | DOL 14 | |
| Oxygen saturation | 86% | 60–70% | 60–70% | 60–70% | 60–70% | |
| Cardiac U/S | Patent foramen ovale and closed ductus arteriosus | Pulmonary hypertension, moderate tricuspid regurgitation | ||||
| Cranial U/S | Normal | Cerebral edema | ||||
| Abdominal U/S | Normal | |||||
| CRP | 3 mg/dL | 13.2 mg/dL | ||||
| WBCs | 13.7/mm3, 38% neutrophils | 14.8/mm3, 84% neutrophils | ||||
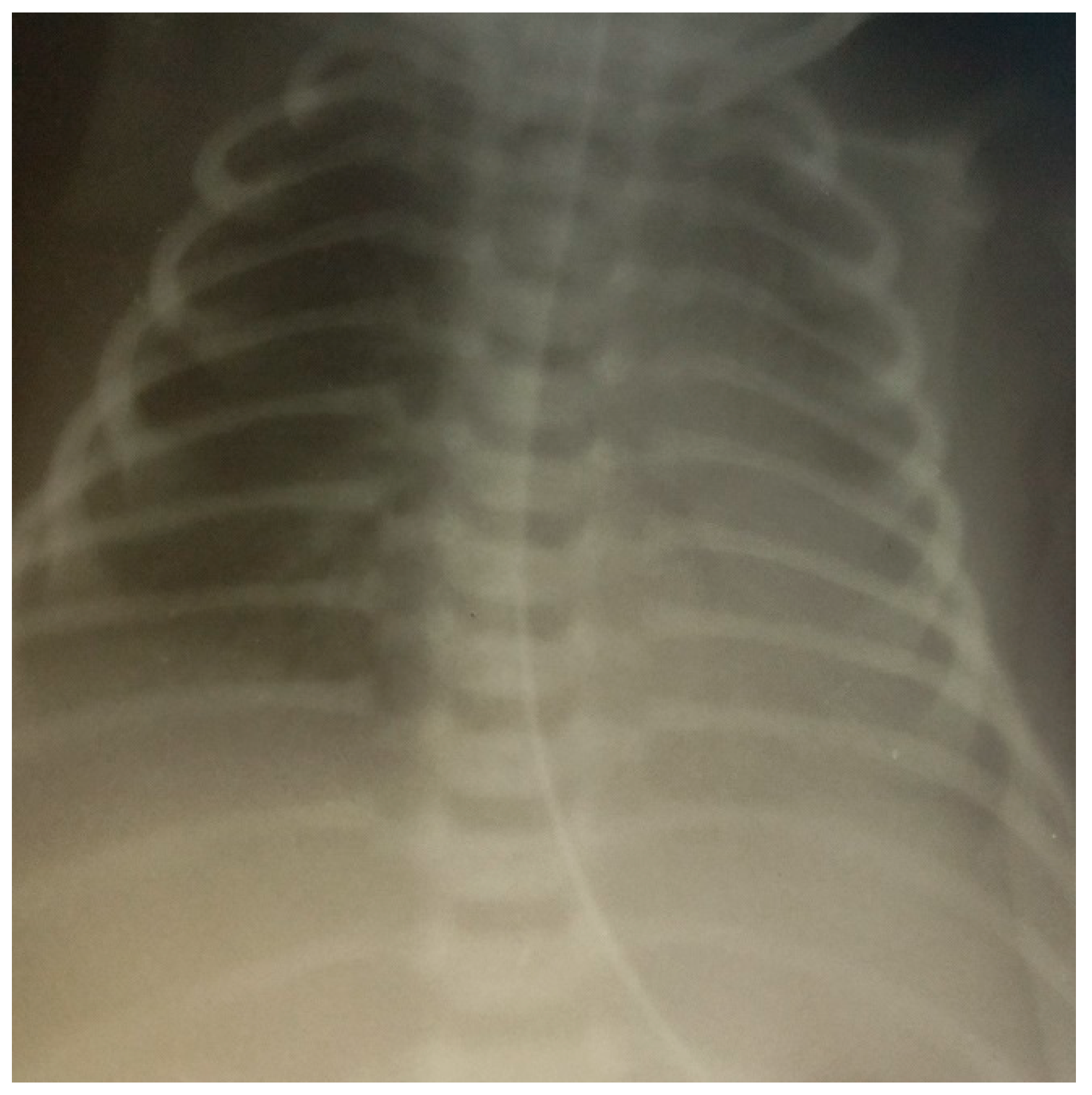
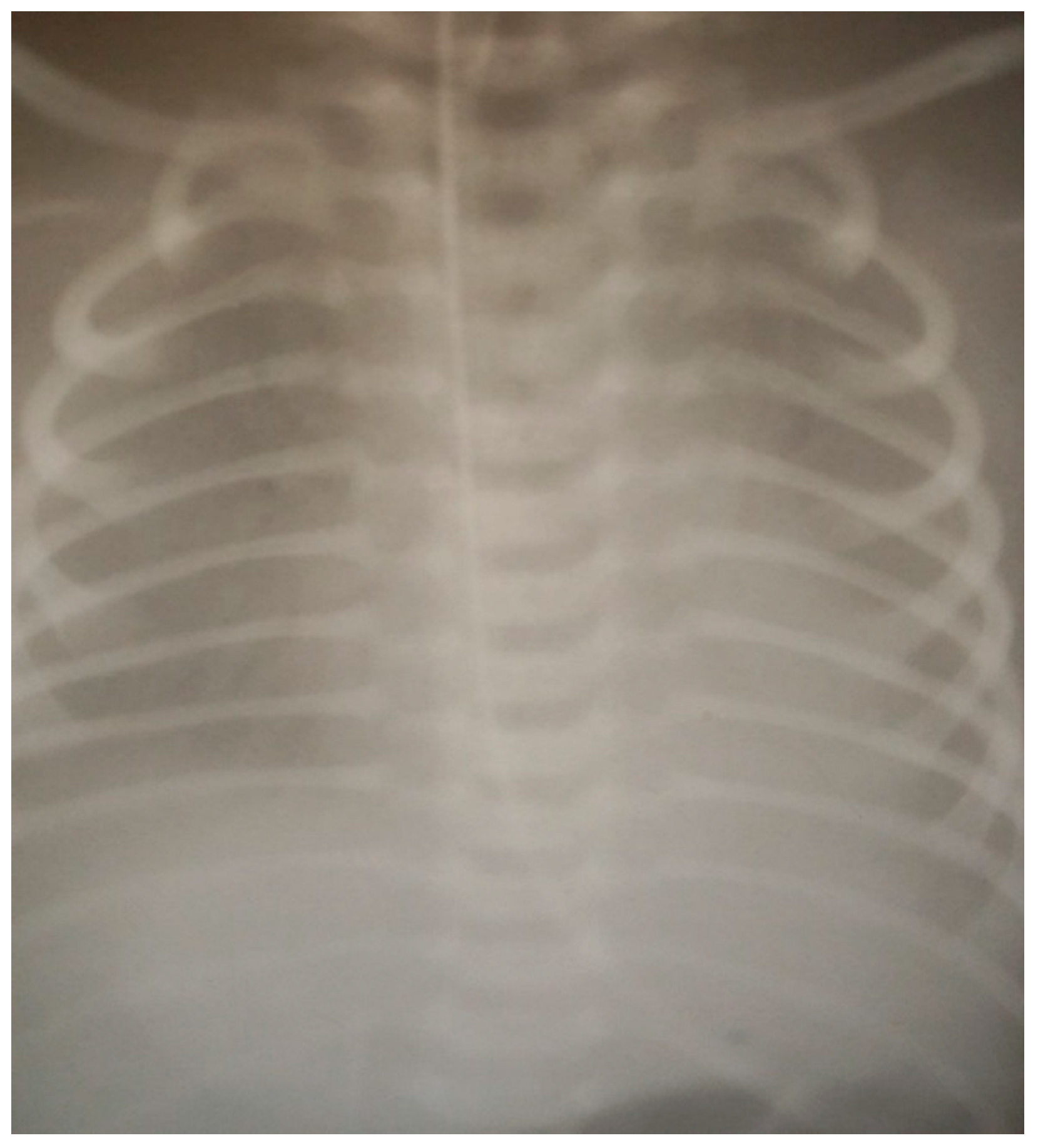
| X-ray | See Figure 1 | See Figure 2 | See Figure 3 | |||
| Lung biopsy | N/A | N/A | N/A | N/A | N/A | Fibrosing alveolitis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabitova, N.K.; Cherezova, I.N.; Arafat, A.; Sadykova, D. Interstitial Lung Disease in Neonates: A Long Road Is Being Paved. Children 2023, 10, 916. https://doi.org/10.3390/children10060916
Gabitova NK, Cherezova IN, Arafat A, Sadykova D. Interstitial Lung Disease in Neonates: A Long Road Is Being Paved. Children. 2023; 10(6):916. https://doi.org/10.3390/children10060916
Chicago/Turabian StyleGabitova, N. Kh., I. N. Cherezova, Ahmed Arafat, and Dinara Sadykova. 2023. "Interstitial Lung Disease in Neonates: A Long Road Is Being Paved" Children 10, no. 6: 916. https://doi.org/10.3390/children10060916