
Natural Course of IQSEC2-Related Encephalopathy: An Italian National Structured Survey
 , , , , , , , , , , , and
, , , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Ethics
2.2. Procedures
2.3. Study Variables
2.4. Clinical Severity
2.4.1. Comparison IQSEC2-Related Encephalopathy with RTT
2.4.2. Genotype–Phenotype relationships
2.4.3. Comparison of IQSEC2 as a Function of Initial RTT Diagnosis vs. RTT
2.5. Sleep Quality
2.6. Quality of Life
2.7. Statistical Analysis
3. Results
3.1. Prevalence Estimates of IQSEC2-Encephalopathy in Italy
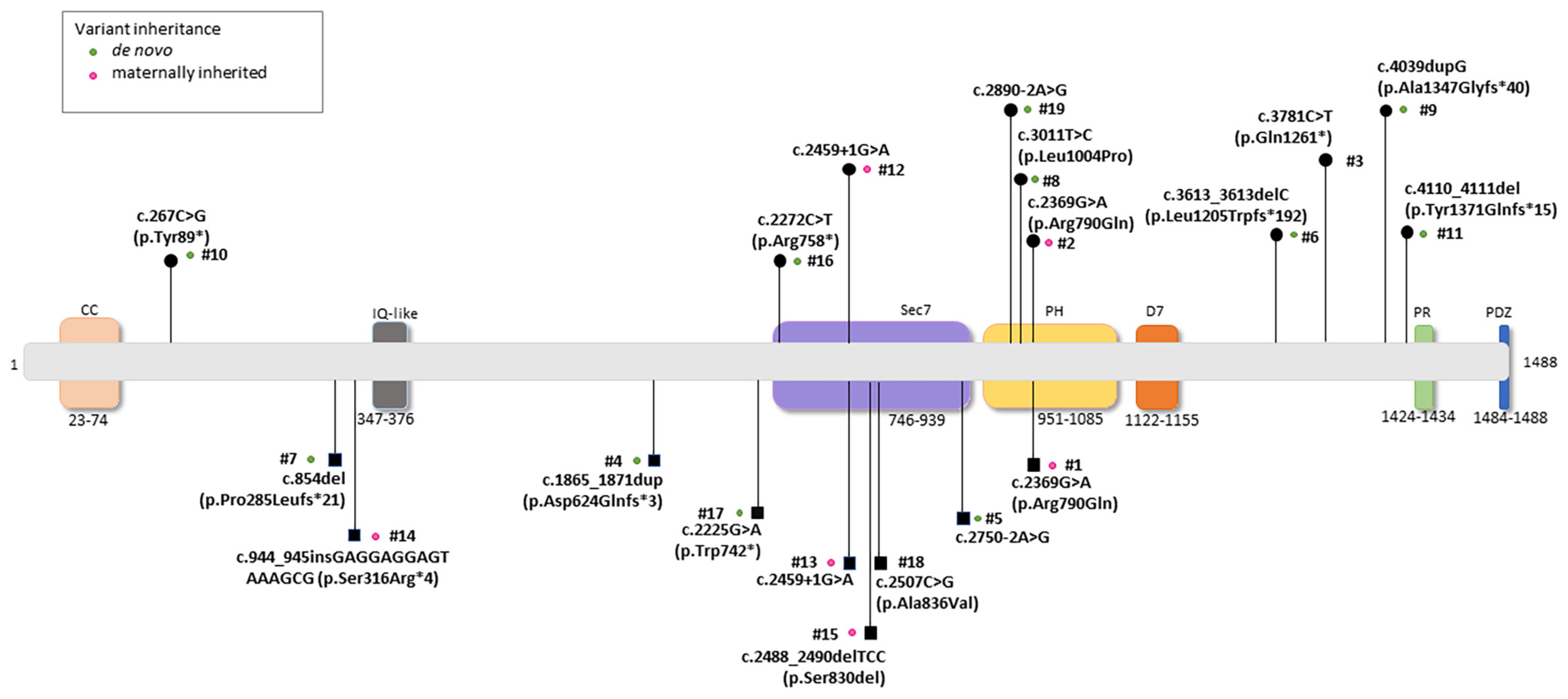
3.2. Molecular Data of the IQSEC2 Population
3.3. IQSEC2-Related Encephalopathy General Overview
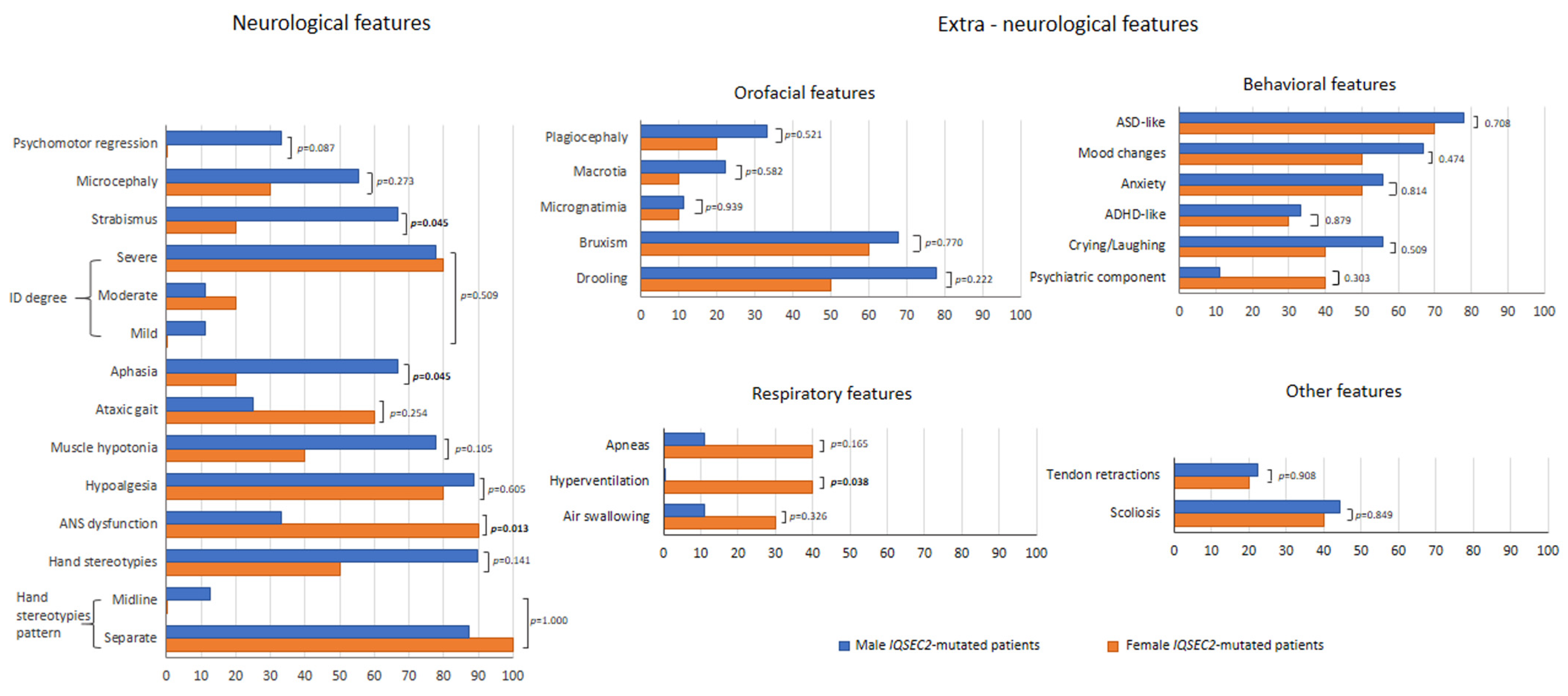
3.4. Perinatal/Neonatal Data, Key Early Developmental Milestones, and Clinical Features as a Function of Gender in the IQSEC2 Population
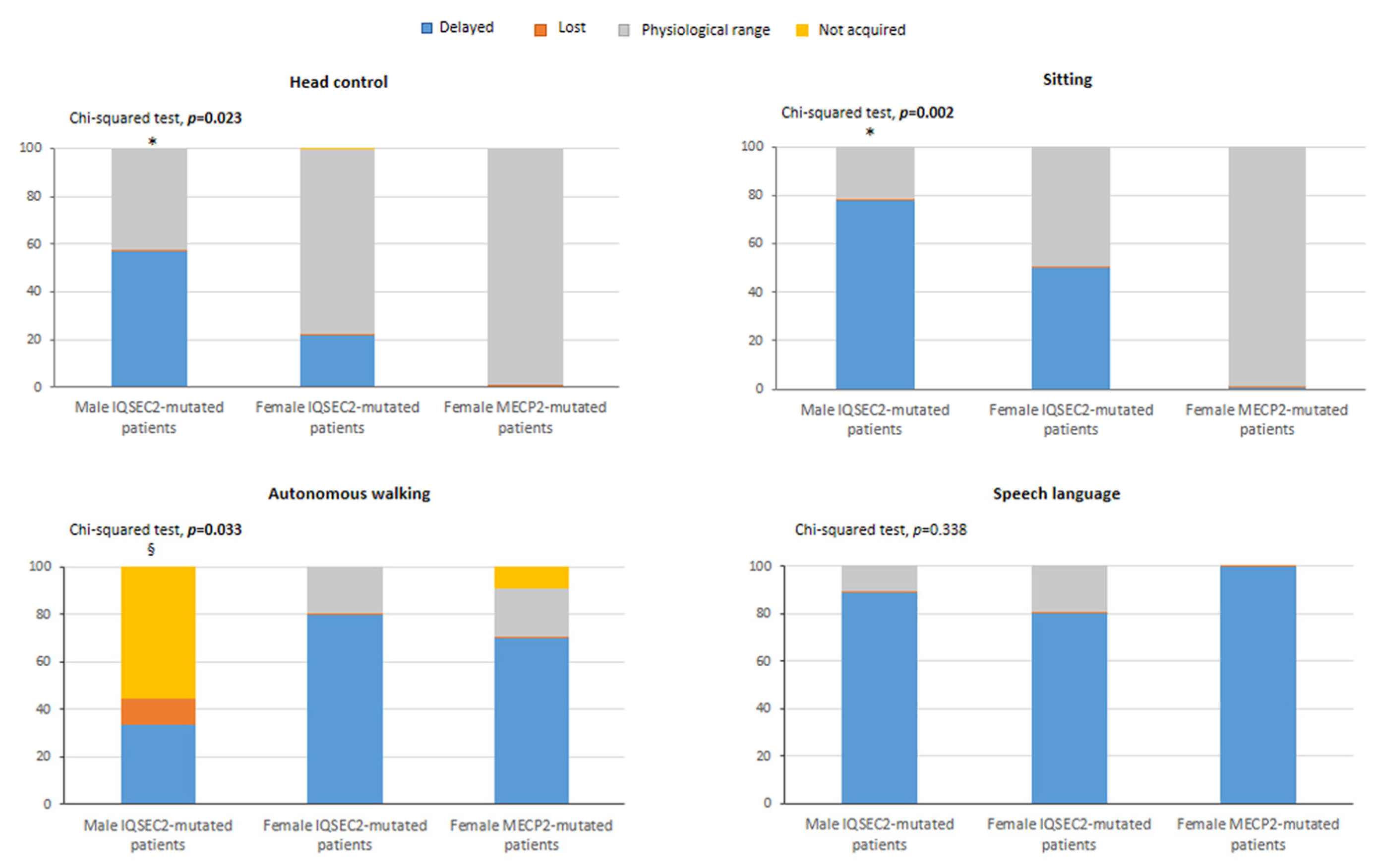
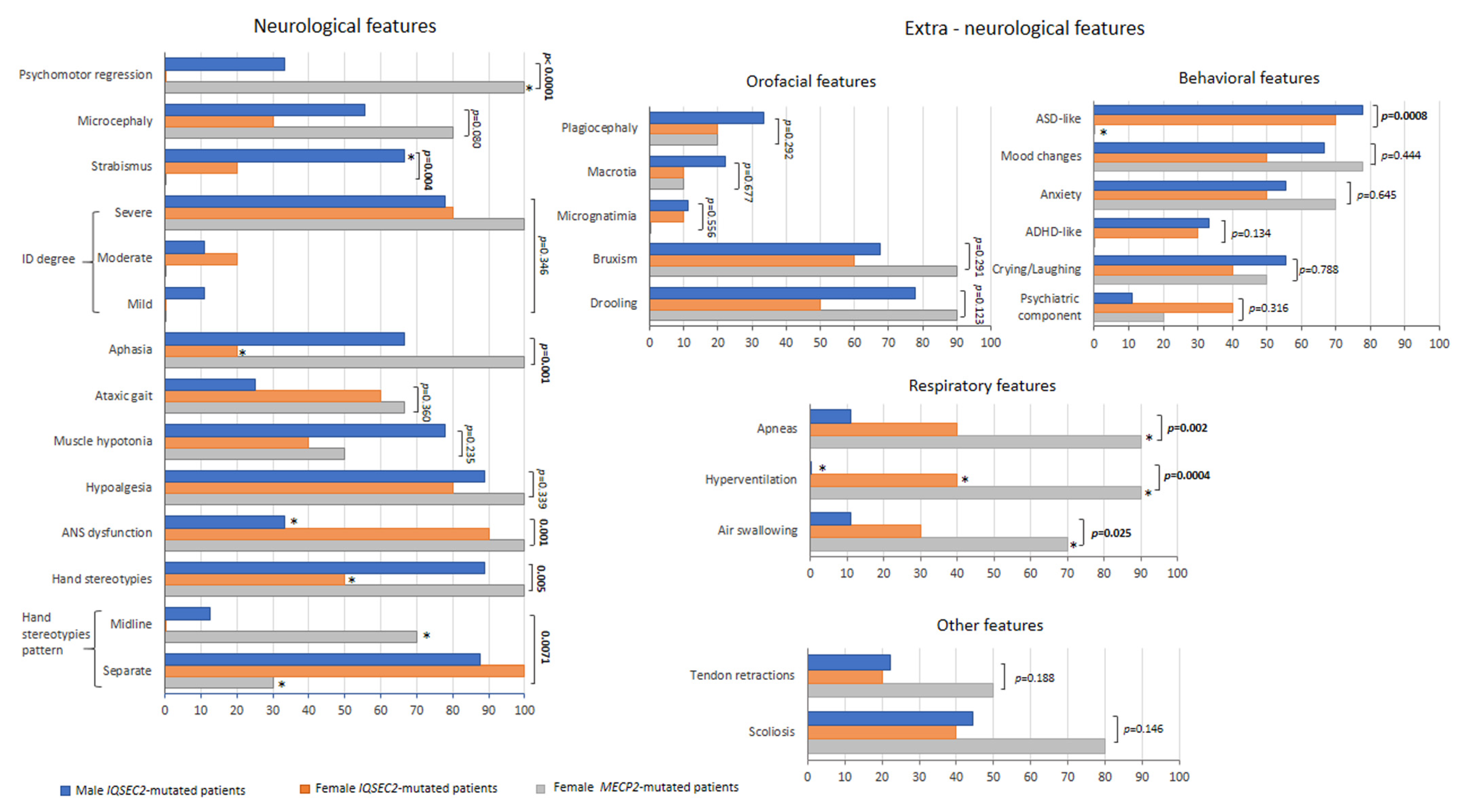
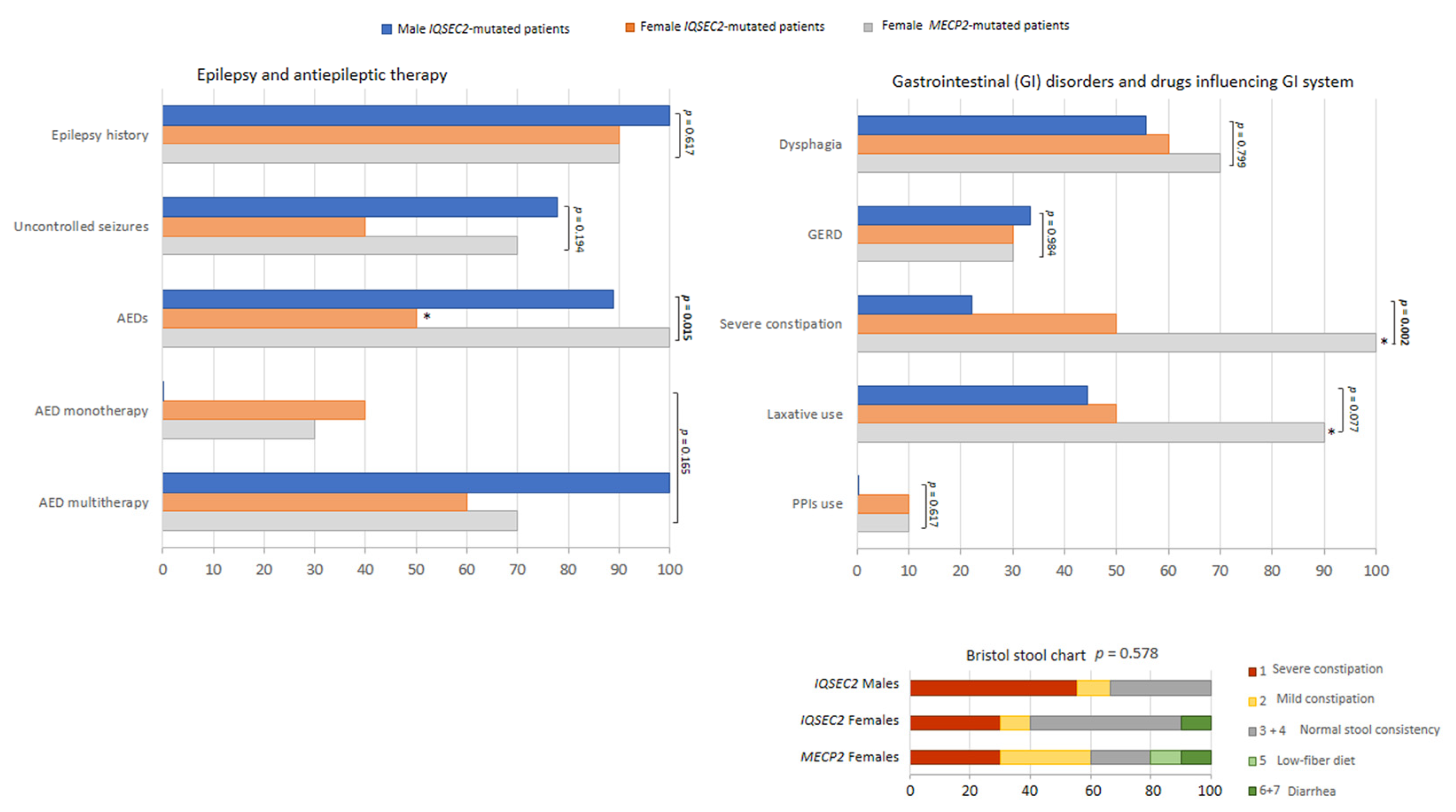
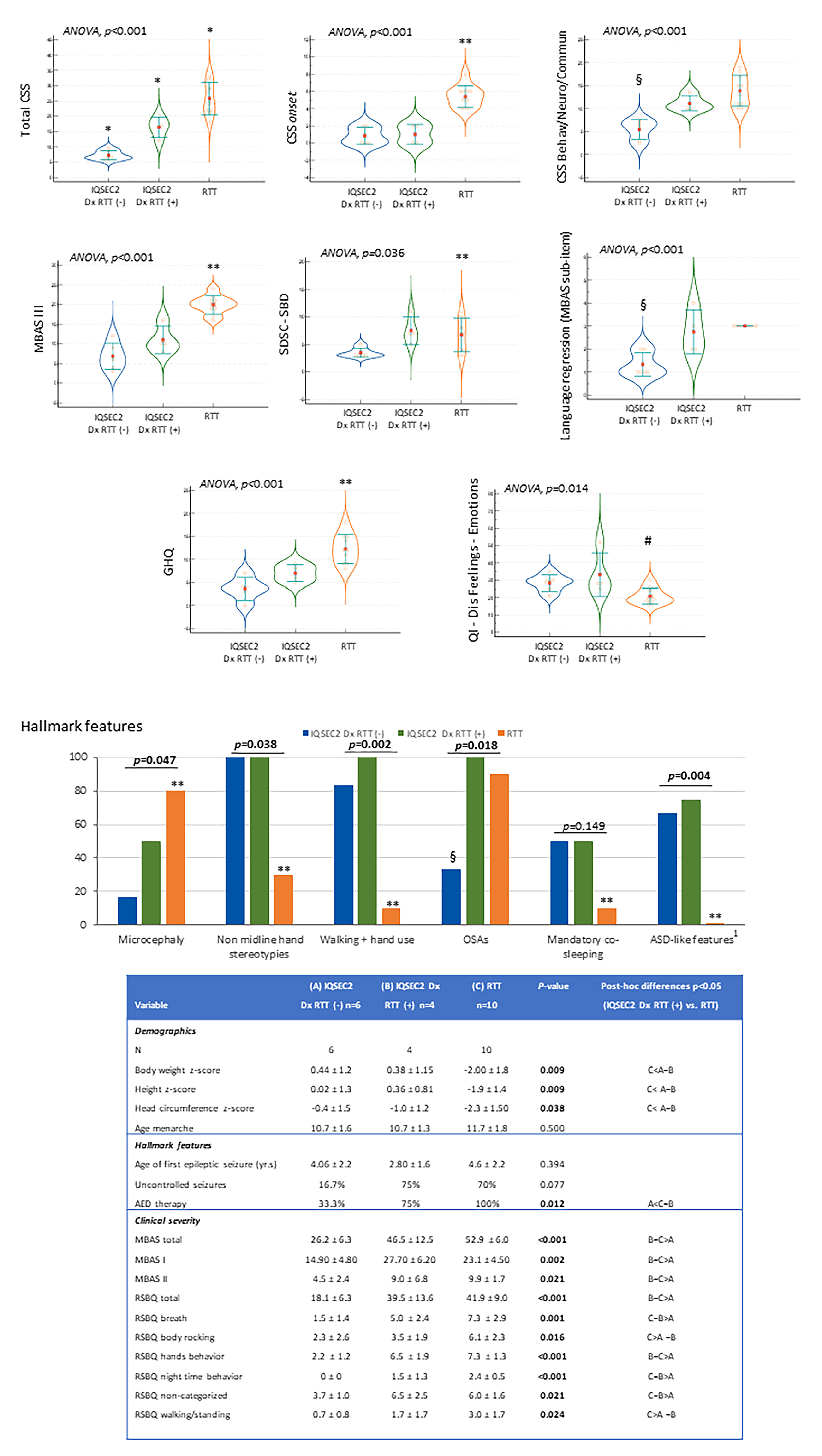
3.5. IQSEC2-Related Encephalopathy vs. RTT: Overlaps and Differences
Clinical Course
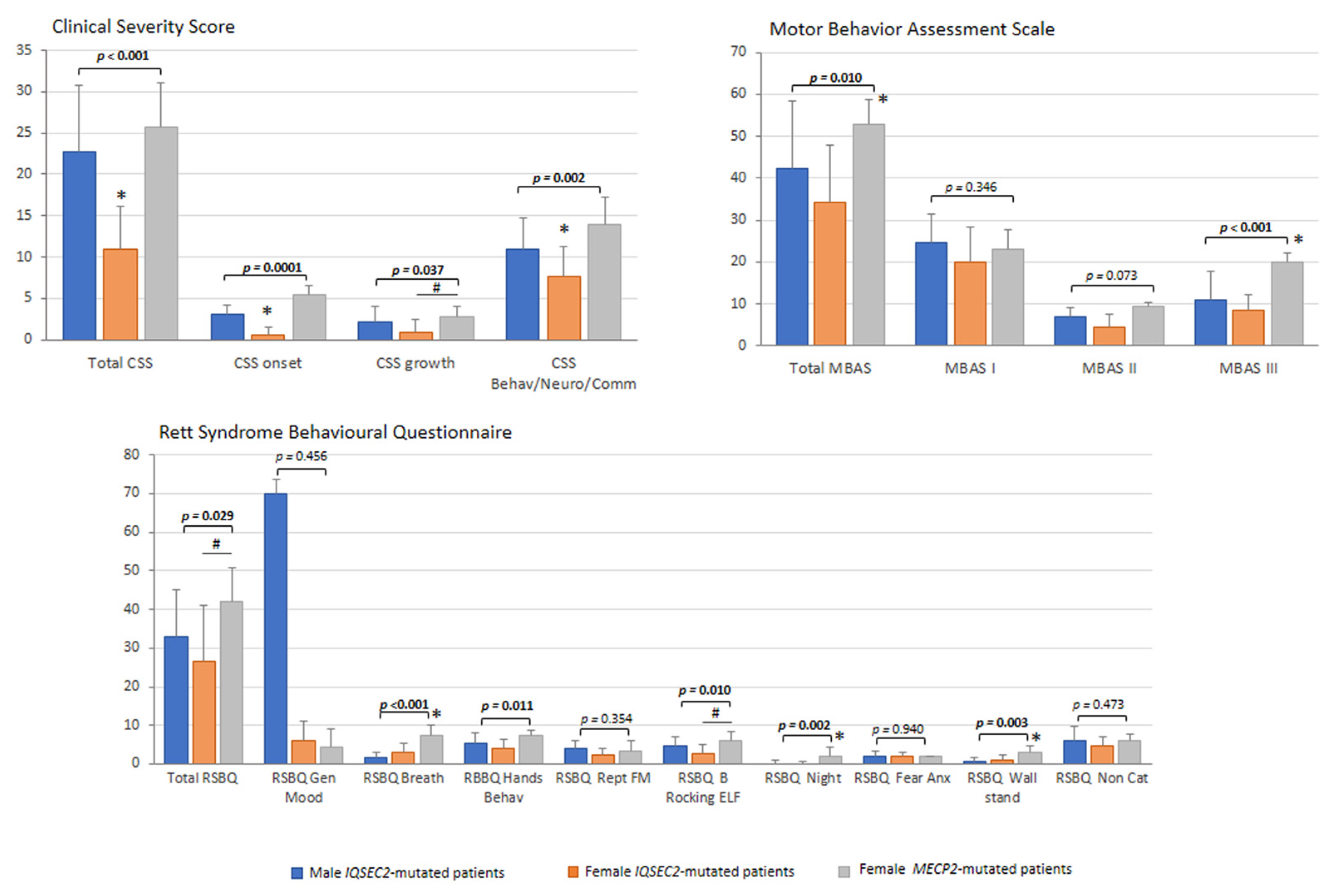
3.6. Illness Severity
3.7. Sleep Disturbance
3.8. Quality of Life
3.9. Miscellaneous Findings
3.10. IQSEC2 Genotype–Phenotype Relationships
3.11. Novel Findings in IQSEC2-Related Encephalopathy
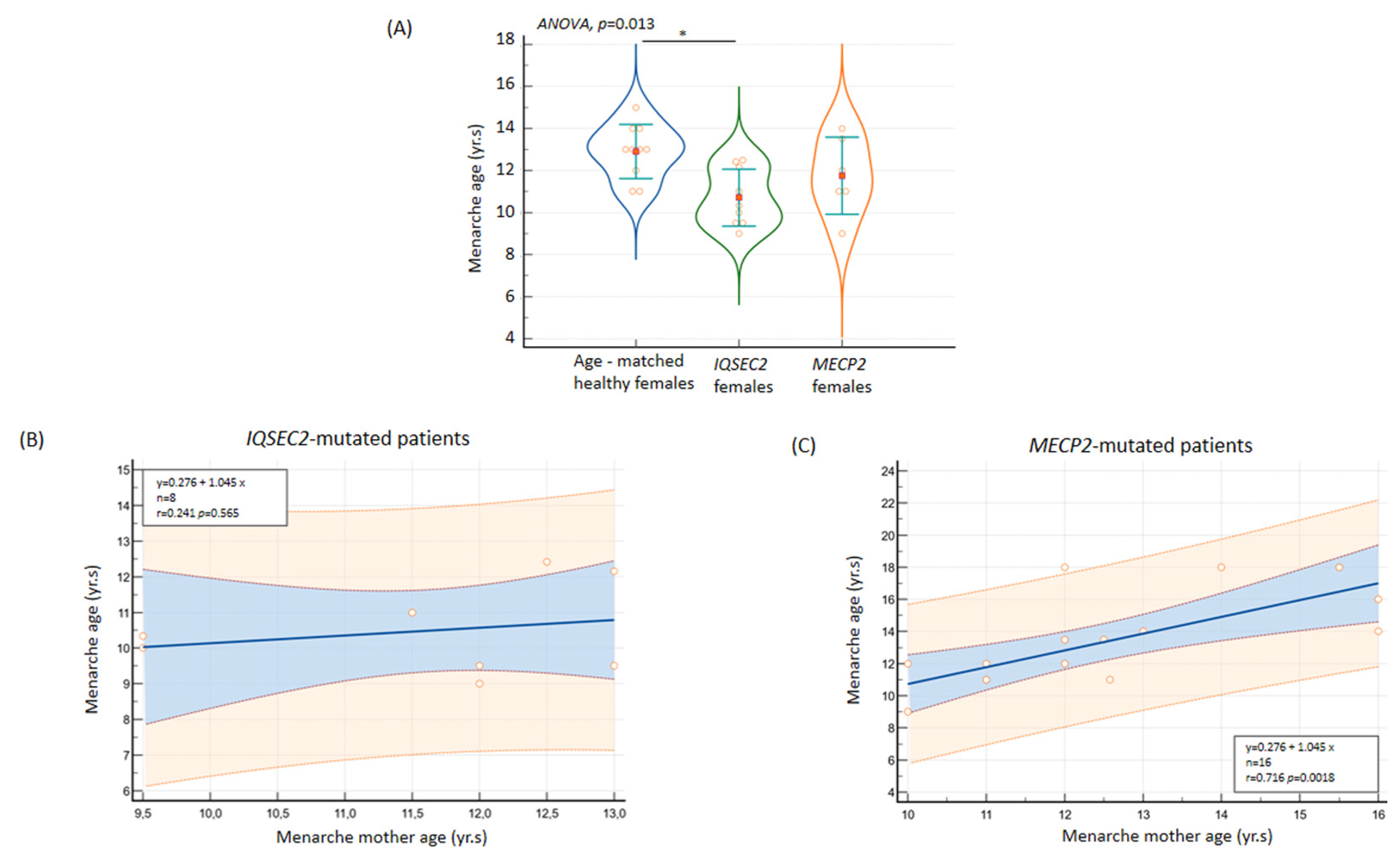
3.11.1. Age at Menarche
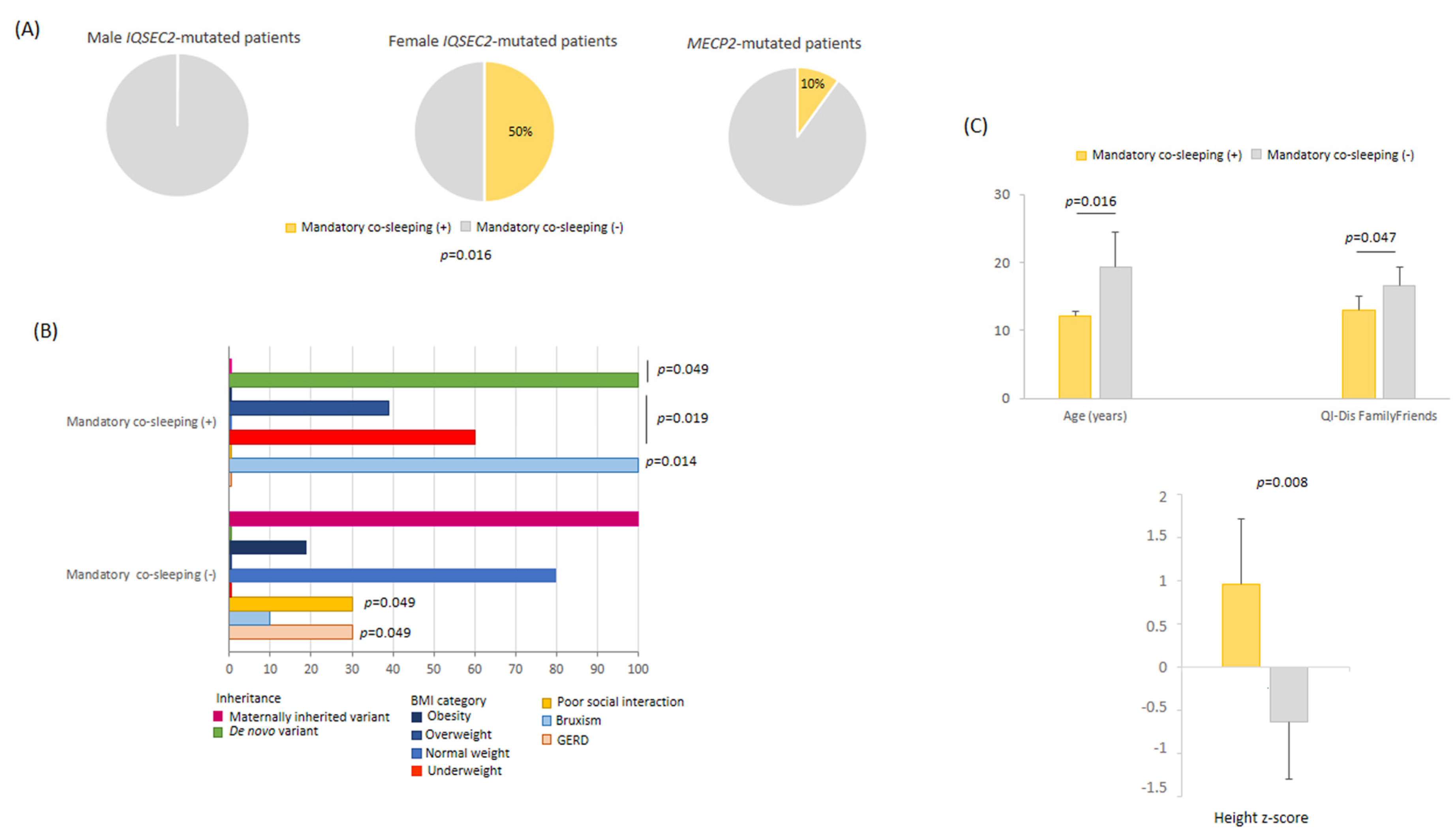
3.11.2. Mandatory Co-Sleeping Behavior
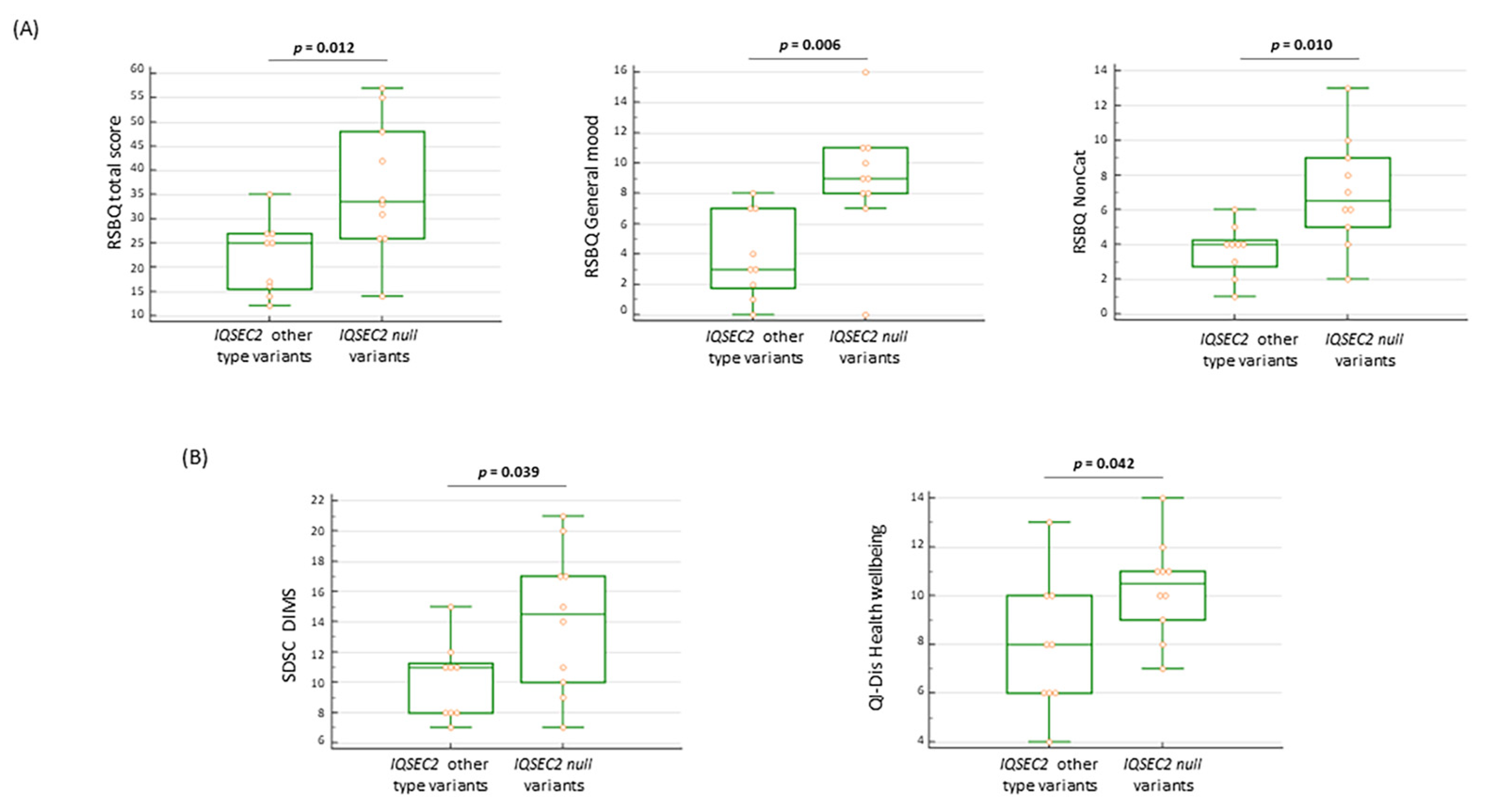
3.11.3. Behavioral and Sleep Features in IQSEC2 Null Variants
3.12. Diagnostic Criteria for RTT in IQSEC2-Related Encephalopathy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander-Bloch, A.F.; McDougle, C.J.; Ullman, Z.; Sweetser, D.A. IQSEC2 and X-linked syndromal intellectual disability. Psychiatr. Genet. 2016, 26, 101–108. [Google Scholar] [CrossRef]
- Lyon, M.F. X-chromosome inactivation and human genetic disease. Acta Paediatr. Suppl. 2002, 91, 107–112. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. X-chromosome inactivation: New insights into cis and trans regulation. Curr. Opin. Genet. Dev. 2015, 31, 57–66. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Shoubridge, C.; Tarpey, P.S.; Abidi, F.; Ramsden, S.L.; Rujirabanjerd, S.; Murphy, J.A.; Boyle, J.; Shaw, M.; Gardner, A.; Proos, A.; et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat. Genet. 2010, 42, 486–488. [Google Scholar] [CrossRef]
- Hinze, S.J.; Jackson, M.R.; Lie, S.; Jolly, L.; Field, M.; Barry, S.C.; Harvey, R.J.; Shoubridge, C. Incorrect dosage of IQSEC2, a known intellectual disability and epilepsy gene, disrupts dendritic spine morphogenesis. Transl. Psychiatry 2017, 7, e1110. [Google Scholar] [CrossRef]
- Elagabani, M.N.; Briševac, D.; Kintscher, M.; Pohle, J.; Köhr, G.; Schmitz, D.; Kornau, H.C. Subunit-selective N-Methyl-d-aspartate (NMDA) receptor signaling through brefeldin A-resistant Arf guanine nucleotide exchange factors BRAG1 and BRAG2 during synapse maturation. J. Biol. Chem. 2016, 291, 9105–9118. [Google Scholar] [CrossRef]
- Brown, J.C.; Petersen, A.; Zhong, L.; Himelright, M.L.; Murphy, J.A.; Walikonis, R.S.; Gerges, N.Z. Bidirectional regulation of synaptic transmission by BRAG1/IQSEC2 and its requirement in long-term depression. Nat. Commun. 2016, 7, 11080. [Google Scholar] [CrossRef]
- IQSEC2-Related Disorder. Available online: https://rarechromo.org/media/information/Chromosome_X/IQSEC2-related%20disorder%20FTNW.pdf (accessed on 5 June 2023).
- Radley, J.A.; O’Sullivan, R.B.G.; Turton, S.E.; Cox, H.; Vogt, J.; Morton, J.; Jones, E.; Smithson, S.; Lachlan, K.; Rankin, J.; et al. Deep phenotyping of 14 new patients with IQSEC2 variants, including monozygotic twins of discordant phenotype. Clin. Genet. 2019, 95, 496–506. [Google Scholar] [CrossRef]
- Lopergolo, D.; Privitera, F.; Castello, G.; Lo Rizzo, C.; Mencarelli, M.A.; Pinto, A.M.; Ariani, F.; Currò, A.; Lamacchia, V.; Canitano, R.; et al. IQSEC2 disorder: A new disease entity or a Rett spectrum continuum? Clin. Genet. 2021, 99, 462–474. [Google Scholar] [CrossRef]
- Mignot, C.; McMahon, A.C.; Bar, C.; Campeau, P.M.; Davidson, C.; Buratti, J.; Nava, C.; Jacquemont, M.L.; Tallot, M.; Milh, M.; et al. IQSEC2-related encephalopathy in males and females: A comparative study including 37 novel patients. Genet. Med. 2019, 21, 1897–1898. [Google Scholar] [CrossRef]
- Shoubridge, C.; Harvey, R.J.; Dudding-Byth, T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum. Mutat. 2019, 40, 5–24. [Google Scholar] [CrossRef]
- Barrie, E.S.; Cottrell, C.E.; Gastier-Foster, J.; Hickey, S.E.; Patel, A.D.; Santoro, S.L.; Alfaro, M.P. Genotype-phenotype correlation: Inheritance and variant-type infer pathogenicity in IQSEC2 gene. Eur. J. Med. Genet. 2020, 63, 103735. [Google Scholar] [CrossRef] [PubMed]
- Heyne, H.O.; Singh, T.; Stamberger, H.; Abou Jamra, R.; Caglayan, H.; Craiu, D.; De Jonghe, P.; Guerrini, R.; Helbig, K.L.; Koeleman, B.P.C.; et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 2018, 50, 1048–1053. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Baladron, B.; Mielu, L.M.; López-Martín, E.; Barrero, M.J.; Lopez, L.; Alvarado, J.I.; Monzón, S.; Varona, S.; Cuesta, I.; Cazorla, R.; et al. Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder. Int. J. Mol. Sci. 2022, 23, 9480. [Google Scholar] [CrossRef]
- Gandomi, S.K.; Farwell Gonzalez, K.D.; Parra, M.; Shahmirzadi, L.; Mancuso, M.J.; Pichurin, P.; Temme, R.; Dugan, S.; Zeng, W.; Tang, S. Diagnostic exome sequencing identifies two novel IQSEC2 mutations associated with X-linked intellectual disability with seizures: Implications for genetic counseling and clinical diagnosis. J. Genet. Couns. 2014, 23, 289–298. [Google Scholar] [CrossRef]
- Tran Mau-Them, F.; Willems, M.; Albrecht, B.; Sanchez, E.; Puechberty, J.; Endele, S.; Schneider, A.; Ruiz Pallares, N.; Missirian, C.; Rivier, F.; et al. Expanding the phenotype of IQSEC2 mutations: Truncating mutations in severe intellectual disability. Eur. J. Hum. Genet. EJHG 2014, 22, 289–292. [Google Scholar] [CrossRef]
- Zipper, R.; Baine, S.D.; Genizi, J.; Maoz, H.; Levy, N.S.; Levy, A.P. Developmental progression of intellectual disability, autism, and epilepsy in a child with an IQSEC2 gene mutation. Clin. Case Rep. 2017, 5, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Accogli, A.; Eric Jarvis, G.; Schiavetto, A.; Lai, L.; Amirali, E.L.; Jimenez Cruz, D.A.; Rivière, J.B.; Trakadis, Y. Psychiatric features and variable neurodevelopment outcome in four females with IQSEC2 spectrum disorder. J. Genet. 2020, 99, 47. [Google Scholar] [CrossRef] [PubMed]
- Levy, N.S.; Borisov, V.; Lache, O.; Levy, A.P. Molecular Insights into IQSEC2 Disease. Int. J. Mol. Sci. 2023, 24, 4984. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef]
- Spagnoli, C.; Fusco, C.; Pisani, F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes 2021, 12, 1157. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Christodoulou, J.; Grimm, A.; Maher, T.; Bennetts, B. RettBASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum. Mutat. 2003, 21, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Borloz, E.; Villard, L.; Roux, J.C. Rett syndrome: Think outside the (skull) box. Fac. Rev. 2021, 10, 59–62. [Google Scholar] [CrossRef]
- Hagberg, B. Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. 2002, 8, 61–65. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. For the RettSearch Consortium (Members listed in the Appendix), Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Boban, S.; Leonard, H.; Wong, K.; Wilson, A.; Downs, J. Sleep disturbances in Rett syndrome: Impact and management including use of sleep hygiene practices. Am. J. Med. Genet. A 2018, 176, 1569–1577. [Google Scholar] [CrossRef]
- Kay, C.; Leonard, H.; Smith, J.; Wong, K.; Downs, J. Genotype and sleep independently predict mental health in Rett syndrome: An observational study. J. Med. Genet. 2023. [Google Scholar] [CrossRef]
- Marcus, C.L.; Carroll, J.L.; McColley, S.A.; Loughlin, G.M.; Curtis, S.; Pyzik, P.; Naidu, S. Polysomnographic characteristics of patients with Rett syndrome. J. Pediatr. 1994, 125, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, M.; Esposito, M.; D’Aniello, A.; Rippa, C.D.; Precenzano, F.; Pascotto, A.; Bravaccio, C.; Elia, M. Polysomnographic findings in Rett syndrome: A case-control study. Sleep Breath. 2013, 17, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Amaddeo, A.; De Sanctis, L.; Arroyo, J.O.; Khirani, S.; Bahi-Buisson, N.; Fauroux, B. Polysomnographic findings in Rett syndrome. Eur. J. Paediatr. Neurol. 2019, 23, 214–221. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Guazzi, G.; Rossi, M.; Ciccoli, L.; Signorini, C.; Leoncini, S.; Tonni, G.; Latini, G.; Valacchi, G.; Hayek, J. Unrecognized lung disease in classic Rett syndrome: A physiologic and high-resolution CT imaging study. Chest 2010, 138, 386–392. [Google Scholar] [CrossRef]
- De Felice, C.; Rossi, M.; Leoncini, S.; Chisci, G.; Signorini, C.; Lonetti, G.; Vannuccini, L.; Spina, D.; Ginori, A.; Iacona, I.; et al. Inflammatory lung disease in Rett syndrome. Mediat. Inflamm. 2014, 2014, 560120. [Google Scholar] [CrossRef]
- Leoncini, S.; Signorini, C.; Boasiako, L.; Scandurra, V.; Hayek, J.; Ciccoli, L.; Rossi, M.; Canitano, R.; De Felice, C. Breathing Abnormalities During Sleep and Wakefulness in Rett Syndrome: Clinical Relevance and Paradoxical Relationship With Circulating Pro-oxidant Markers. Front. Neurol. 2022, 13, 833239. [Google Scholar] [CrossRef]
- Bebbington, A.; Anderson, A.; Ravine, D.; Fyfe, S.; Pineda, M.; De Klerk, N.; Ben-Zeev, B.; Yatawara, N.; Percy, A.; Kaufmann, W.E.; et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008, 70, 868–875. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef]
- Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt, R.; Standridge, S.; Lane, J.; Dinkel, T.; Jones, M.; et al. Multisystem comorbidities in classic Rett syndrome: A scoping review. BMJ Paediatr. Open 2020, 4, e000731. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Litvak, V.; Kipnis, J. Unexpected cellular players in Rett syndrome pathology. Neurobiol. Dis. 2016, 92 Pt A, 64–71. [Google Scholar] [CrossRef]
- Motil, K.J.; Caeg, E.; Barrish, J.O.; Geerts, S.; Lane, J.B.; Percy, A.K.; Annese, F.; McNair, L.; Skinner, S.A.; Lee, H.S.; et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 292–298. [Google Scholar] [CrossRef]
- Frankel, E.; Podder, A.; Sharifi, M.; Pillai, R.; Belnap, N.; Ramsey, K.; Dodson, J.; Venugopal, P.; Brzezinski, M.; Llaci, L.; et al. Genetic and Protein Network Underlying the Convergence of Rett-Syndrome-like (RTT-L) Phenotype in Neurodevelopmental Disorders. Cells 2023, 12, 1437. [Google Scholar] [CrossRef]
- Gonzalez, J.N.; Goldman, S.; Carter, M.T.; Bain, J.M. Rett-like Phenotypes in HNRNPH2-Related Neurodevelopmental Disorder. Genes 2023, 4, 1154. [Google Scholar] [CrossRef] [PubMed]
- Associazione AMA.le IQSEC2. Available online: https://amaleiqsec2.com/ (accessed on 1 April 2023).
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdò, S.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- ClinVar Tool. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 5 June 2023).
- Cacciari, E.; Milani, S.; Balsamo, A.; Spada, E.; Bona, G.; Cavallo, L.; Cerutti, F.; Gargantini, L.; Greggio, N.; Tonini, G.; et al. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J. Endocrinol. Investig. 2006, 29, 581–593. [Google Scholar] [CrossRef]
- Tarquinio, D.C.; Motil, K.J.; Hou, W.; Lee, H.S.; Glaze, D.G.; Skinner, S.A.; Neul, J.L.; Annese, F.; McNair, L.; Barrish, J.O.; et al. Growth failure and outcome in Rett syndrome: Specific growth references. Neurology 2012, 79, 1653–1661. [Google Scholar] [CrossRef]
- Rare Diseases: Signs and Symptoms. Available online: https://www.orpha.net/consor/cgi-bin/Disease_HPOTerms.php?lng=EN (accessed on 30 June 2023).
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- FitzGerald, P.M.; Jankovic, J.; Percy, A.K. Rett syndrome and associated movement disorders. Mov. Disord. 1990, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mount, R.H.; Charman, T.; Hastings, R.P.; Reilly, S.; Cass, H. The Rett Syndrome Behaviour Questionnaire (RSBQ): Refining the behavioural phenotype of Rett syndrome. J. Child. Psychol. Psychiatry 2002, 43, 1099–1110. [Google Scholar] [CrossRef]
- Motil, K.J.; Khan, N.; Coon, J.L.; Barrish, J.O.; Suter, B.; Pehlivan, D.; Schultz, R.J.; Glaze, D.G. Gastrointestinal Health Questionnaire for Rett Syndrome: Tool Development. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Heaton, K.W. Stool form scale as a useful guide to intestinal transit time. Scand. J. Gastroenterol. 1997, 32, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Bruni, O.; Ottavianio, S.; Guidetti, V.; Romoli, M.; Innocenzi, M.; Cortesi, F.; Giannotti, F. The Sleep Disturbance Scale for Children (SDSC): Construction and validation of an instrument to evaluate sleep disturbances in childhood and adolescence. J. Sleep Res. 1996, 5, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.; Jacoby, P.; Leonard, H.; Epstein, A.; Murphy, N.; Davis, E.; Reddihough, D.; Whitehouse, A.; Williams, K. Psychometric properties of the Quality of Life Inventory-Disability (QI-Disability) measure. Qual. Life Res. 2019, 28, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.; Williams, K.; Reddihough, D.; Murphy, N.; Leonard, H.; Whitehouse, A.; Jacoby, P.; Downs, J. Content validation of the Quality of Life Inventory-Disability. Child. Care Health Dev. 2019, 45, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Piras, G.N.; Bozzola, M.; Bianchin, L.; Bernasconi, S.; Bona, G.; Lorenzoni, G.; Buzi, F.; Rigon, F.; Tonini, G.; De Sanctis, V.; et al. The levelling-off of the secular trend of age at menarche among Italian girls. Heliyon 2020, 6, e04222. [Google Scholar] [CrossRef] [PubMed]
- Roderick Little, J.A.; Donald Rubin, B. Statistical Analysis with Missing Data, 2nd ed.; Wiley Series in Probability and Statistics; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 79, pp. 27–29. [Google Scholar]
- Italian Age Pyramid. Available online: https://www.tuttitalia.it/statistiche/popolazione-eta-sesso-stato-civile-2022/ (accessed on 5 June 2023).
- May, D.; Kponee-Shovein, K.; Mahendran, M.; Downes, N.; Sheng, K.; Lefebvre, P.; Cheng, W.Y. Epidemiology and patient journey of Rett syndrome in the United States: A real-world evidence study. BMC Neurol. 2023, 23, 141. [Google Scholar] [CrossRef]
- Petriti, U.; Dudman, D.C.; Scosyrev, E.; Lopez-Leon, S. Global prevalence of Rett syndrome: Systematic review and meta-analysis. Syst. Rev. 2023, 12, 5. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered gut microbiota in Rett syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef]
- Tieder, J.S.; Bonkowsky, J.L.; Etzel, R.A.; Franklin, W.H.; Gremse, D.A.; Herman, B.; Katz, E.S.; Krilov, L.R.; Merritt, J.L., 2nd; Norlin, C.; et al. Brief Resolved Unexplained Events (Formerly Apparent Life-Threatening Events) and Evaluation of Lower-Risk Infants. Pediatrics 2016, 137, e20160590. [Google Scholar] [CrossRef]
- National Demographic Indicators (Mother’s Age at the First Childbirth). Available online: https://demo.istat.it/tavole/?t=indicatori&l=en (accessed on 5 July 2023).
- Brunet, T.; Jech, R.; Brugger, M.; Kovacs, R.; Alhaddad, B.; Leszinski, G.; Riedhammer, K.M.; Westphal, D.S.; Mahle, I.; Mayerhanse, K.; et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 2021, 100, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.A.; Goriely, A. The impact of paternal age on new mutations and disease in the next generation. Fertil. Steril. 2022, 118, 1001–1012. [Google Scholar] [CrossRef]
- National Demographic Indicators (Father’s Age at the First Childbirth). Available online: http://dati-giovani.istat.it/Index.aspx?QueryId=10979# (accessed on 5 July 2023).
- Rogers, E.; Jada, R.; Schragenheim-Rozales, K.; Sah, M.; Cortes, M.; Florence, M.; Levy, N.S.; Moss, R.; Walikonis, R.S.; Palty, R.; et al. An IQSEC2 Mutation Associated with Intellectual Disability and Autism Results in Decreased Surface AMPA Receptors. Front. Mol. Neurosci. 2019, 12, 43. [Google Scholar] [CrossRef]
- Jackson, M.R.; Loring, K.E.; Homan, C.C.; Thai, M.H.; Määttänen, L.; Arvio, M.; Jarvela, I.; Shaw, M.; Gardner, A.; Gecz, J.; et al. Heterozygous loss of function of IQSEC2/Iqsec2 leads to increased activated Arf6 and severe neurocognitive seizure phenotype in females. Life Sci. Alliance 2019, 2, e201900386. [Google Scholar] [CrossRef] [PubMed]
- Levy, N.S.; Umanah, G.K.E.; Rogers, E.J.; Jada, R.; Lache, O.; Levy, A.P. IQSEC2-Associated Intellectual Disability and Autism. Int. J. Mol. Sci. 2019, 20, 3038. [Google Scholar] [CrossRef]
- Sah, M.; Shore, A.N.; Petri, S.; Kanber, A.; Yang, M.; Weston, M.C.; Frankel, W.N. Altered excitatory transmission onto hippocampal interneurons in the IQSEC2 mouse model of X-linked neurodevelopmental disease. Neurobiol. Dis. 2020, 137, 104758. [Google Scholar] [CrossRef] [PubMed]
- IQSEC2 and Pain Sensitivity. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IQSEC2 (accessed on 14 July 2023).
- National Epidemiological Data on Breastfeeding (Istituto Superiore di Sanità). Available online: https://www.epicentro.iss.it/en/breastfeeding/epid-italy (accessed on 30 October 2022).
- Thomas, R.H.; Berkovic, S.F. The hidden genetics of epilepsy-a clinically important new paradigm. Nat. Rev. Neurol. 2014, 10, 283–292. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Raga, S.; Specchio, N.; Rheims, S.; Wilmshurst, J.M. Developmental and epileptic encephalopathies: Recognition and approaches to care. Epileptic Disord. 2021, 23, 40–52. [Google Scholar] [CrossRef]
- Helbig, I.; Heinzen, E.L.; Mefford, H.C.; ILAE Genetics Commission. Primer Part 1-The building blocks of epilepsy genetics. Epilepsia 2016, 57, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Tzschach, A.; Grasshoff, U.; Beck-Woedl, S.; Dufke, C.; Bauer, C.; Kehrer, M.; Evers, C.; Moog, U.; Oehl-Jaschkowitz, B.; Di Donato, N.; et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 2015, 23, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Helm, B.M.; Powis, Z.; Prada, C.E.; Casasbuenas-Alarcon, O.L.; Balmakund, T.; Schaefer, G.B.; Kahler, S.G.; Kaylor, J.; Winter, S.; Zarate, Y.A.; et al. The role of IQSEC2 in syndromic intellectual disability: Narrowing the diagnostic odyssey. Am. J. Med. Genet. A 2017, 173, 2814–2820. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. Fundamental Concepts in Genetics: Genetics and the Understanding of Selection. Nat. Rev. Genet. 2009, 10, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Moey, C.; Hinze, S.J.; Brueton, L.; Morton, J.; McMullan, D.J.; Kamien, B.; Barnett, C.P.; Brunetti-Pierri, N.; Nicholl, J.; Gecz, J.; et al. Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet. 2016, 24, 373–380. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef]
- SMARCC1 Gene Description. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=SMARCC1 (accessed on 14 July 2023).
- Bastida-Pozuelo, M.F.; Meltzer, L.J.; Sánchez-Ortuño, M.M. Cosleeping and Behavioral Sleep Problems in School-aged Children with Neurodevelopmental and Mental Health Disorders. Arch. Psychiatr. Nurs. 2018, 32, 483–487. [Google Scholar] [CrossRef]
- Cohen, S.; Conduit, R.; Lockley, S.W.; Rajaratnam, S.M.; Cornish, K.M. The relationship between sleep and behavior in autism spectrum disorder (ASD): A review. J. Neurodev. Disord. 2014, 6, 44. [Google Scholar] [CrossRef]
- Al Lihabi, A. A literature review of sleep problems and neurodevelopment disorders. Front. Psychiatry 2023, 14, 1122344. [Google Scholar] [CrossRef]
- Fu, L.Y.; Colson, E.R.; Corwin, M.J.; Moon, R.Y. Infant sleep location: Associated maternal and infant characteristics with sudden infant death syndrome prevention recommendations. J. Pediatr. 2008, 153, 503–508. [Google Scholar] [CrossRef]
- Colson, E.R.; Willinger, M.; Rybin, D.; Heeren, T.; Smith, L.A.; Lister, G.; Corwin, M.J. Trends and factors associated with infant bed sharing, 1993–2010: The National Infant Sleep Position Study. JAMA Pediatr. 2013, 167, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hubbard, J.A.; Fabes, R.A.; Adam, J.B. Sleep disturbances and correlates of children with autism spectrum disorders. Child. Psychiatry Hum. Dev. 2006, 37, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Argente, J.; Dunkel, L.; Kaiser, U.B.; Latronico, A.C.; Lomniczi, A.; Soriano-Guillén, L.; Tena-Sempere, M. Molecular basis of normal and pathological puberty: From basic mechanisms to clinical implications. Lancet Diabetes Endocrinol. 2023, 11, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Mucci, A.; Clemente, E. The Role of Genetics in Central Precocious Puberty: Confirmed and Potential Neuroendocrine Genetic and Epigenetic Contributors and Their Interactions with Endocrine Disrupting Chemicals (EDCs). Endocrines 2022, 3, 433–451. [Google Scholar] [CrossRef]
- IQSEC2 Gene Related Symptoms and Diseases. Available online: https://www.mendelian.co/genes/iqsec2 (accessed on 14 July 2023).
- Killian, J.T.; Lane, J.B.; Cutter, G.R.; Skinner, S.A.; Kaufmann, W.E.; Tarquinio, D.C.; Glaze, D.G.; Motil, K.J.; Neul, J.L.; Percy, A.K. Pubertal development in Rett syndrome deviates from typical females. Pediatr. Neurol. 2014, 51, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.I.E.; Bergman, P.; Hagey, D.W. Estimating the number of diseases—The concept of rare, ultra-rare, and hyper-rare. iScience 2022, 25, 104698. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | IQSEC2-Mutated Whole Population (19) | IQSEC2-Mutated Males (9) | IQSEC2-Mutated Females (10) | Comparison between IQSEC2 M vs. F p-Value |
|---|---|---|---|---|
| Demographics | ||||
| N | 19 | 9 | 10 | |
| Age (years) | 13.7 ± 7.3 (range 2.3–34) | 12.7 [3.3–15.8] | 13.1 [12.1–19.4] | 0.094 |
| Age category | ||||
| Pediatric | 9 (47.4%) | 5 (55.6%) | 4 (40%) | 0.588 |
| Adolescent | 6 (31.6%) | 3 (33.3%) | 3 (30%) | |
| Adult | 4 (21.1%) | 1 (1.11%) | 3 (30%) | |
| Genetic diagnosis ≤ 2 years | 1/19 (5.3%) | 1/9 (11.1%) | 0 (0%) | 0.292 |
| Body weight z-score | −0.18 ± 1.37 | −0.86 ± 1.37 | 0.42 ± 1.11 | 0.040 |
| Height z-score | −0.15 ± 1.14 | −0.49 ± 1.17 | −0.16 ± 1.08 | 0.223 |
| Head circumference z-score | −0.92 ± 1.03 | −1.22 ± 0.79 | −0.65 ± 1.17 | 0.232 |
| BMI z-score | −0.29 ± 1.47 | −0.87 ± 1.37 | 0.24 ± 1.42 | 0.101 |
| Perinatal/neonatal data | ||||
| Gestational age (weeks) | 38.5 ± 2.7 | 40 [38.0–40.2] | 38 [37.0–40.0] | 0.318 |
| Prematurity | 3 (15.8%) | 1 (11.1%) | 2 (20%) | 1.000 |
| Adverse perinatal events | 6 (31.6%) | 2 (22.2%) | 4 (40%) | 0.418 |
| Neonatal birth weight (g) | 3120.0 ± 513.4 | 3180.0 ± 605.2 | 3066.8 ± 441.0 | 0.452 |
| Neonatal birth weight z-score | 0.03 ± 1.11 | −0.21 ± 0.93 | 0.26 ± 1.25 | 0.367 |
| Birth weight category | ||||
| AGA | 14 (73.7%) | 7 (77.8%) | 7 (70%) | 0.869 |
| LGA | 3 (15.8%) | 1 (11.1%) | 2 (20%) | |
| SGA | 2 (10.5%) | 1 (11.1%) | 1 (10%) | |
| Delivery mode | ||||
| SVD | 13 (68.4%) | 6 (66.7%) | 7 (70%) | 0.219 |
| ELCS | 2 (10.5%) | 2 (22.2%) | 0 (0%) | |
| EMCS | 4 (21.1%) | 1 (11.1%) | 3 (30%) | |
| Breastfeeding | 11 (57.9%) | 4 (44.4%) | 7 (70%) | 0.273 |
| Breastfeeding duration (months) | 10.8 ± 10.6 | 8.5 [6.0–16.5] | 12 [6.1–12.0] | 0.719 |
| Key early developmental milestones | ||||
| Head control | ||||
| Delayed | 6 (37.5%) | 4 (57.1%) | 2 (22.2%) | 0.166 |
| Physiological range | 10 (62.5%) | 3 (42.9%) | 7 (77.8%) | |
| Autonomous sitting | ||||
| Delayed | 12 (63.2%) | 7 (77.8%) | 5 (50%) | 0.222 |
| Physiological range | 7 (36.8%) | 2 (22.2%) | 5 (50%) | |
| Autonomous walking | ||||
| Delayed | 11 (57.9%) | 3 (33.3%) | 8 (80%) | 0.017 |
| Lost | 1 (5.3%) | 1 (11.1%) | 0 (0%) | |
| Physiological range | 2 (10.5%) | 0 (0%) | 2 (20%) | |
| Not acquired | 5 (26.3%) | 5 (55.6%) | 0 (0%) | |
| Speech | ||||
| Delayed or regressed | 16 (84.2%) | 8 (88.9%) | 8 (80%) | 0.606 |
| Physiological range | 3 (15.8%) | 1 (11.1%) | 2 (20%) |
| Variable | A IQSEC2-Mutated Males | B IQSEC2-Mutated Females | C MECP2-Mutated Females | Comparison between Groups (p-Value) | Post Hoc Differences p < 0.05 |
|---|---|---|---|---|---|
| Demographics | |||||
| N | 9 | 9 | 10 | ||
| Age (years) | 10.7 ± 6.6 | 16.4 ± 7.2 | 15.5 ± 7.7 | 0.210 | |
| Age category | |||||
| Pediatric | 9 (47.4%) | 5 (%) | 4 (%) | 0.588 | |
| Adolescent | 6 (31.6%) | 3 (%) | 3 (%) | ||
| Adult | 4 (21.1%) | 1 (%) | 3 (%) | ||
| Genetic diagnosis ≤ 2 years | |||||
| 2 (22.2%) | 0 (0%) | 8 (80%) | 0.0005 | C > A ~ B | |
| Body weight z-score | −0.86 ± 1.37 | 0.42 ± 1.11 | −2.00 ± 1.76 | 0.004 | C < B ~ A |
| Height z-score | −0.49 ± 1.17 | −0.16 ± 1.08 | −1.90 ± 1.44 | 0.003 | C < B ~ A |
| Head circumference z-score | −1.23 ± 0.79 | −0.65 ± 1.17 | −2.3 ± 1.5 | 0.015 | C < B ~ A |
| BMI z-score | −0.87 ± 1.37 | 0.24 ± 1.42 | −1.26 ± 1.41 | 0.062 | |
| Perinatal/neonatal data | |||||
| Gestational age (weeks) | 40 [38.0–40.2] | 38 [37.0–40.0] | 40 [40.0–40.0] | 0.250 | |
| Prematurity | 1 (11.1%) | 2 (20%) | 0 (0%) | 0.339 | |
| Adverse perinatal events | 2 (22.2%) | 4 (40%) | 1 (10%) | 0.289 | |
| Neonatal birth weight (g) | 3180.0 ± 605.2 | 3066.8 ± 441.0 | 3187.0 ± 223.4 | 0.795 | |
| Neonatal birth weight z-score | −0.21 ± 0.93 | 0.26 ± 1.25 | −0.34 ± 0.36 | 0.379 | |
| Birth weight classification | |||||
| AGA | 7 (77.8%) | 7 (70%) | 9 (90%) | 0.698 | |
| LGA | 1 (11.1%) | 2 (20%) | 0 (0%) | ||
| SGA | 1 (11.1%) | 1 (10%) | 1 (10%) | ||
| Delivery mode | |||||
| SVD | 6 (66.7%) | 7 (70%) | 8 (80%) | 0.249 | |
| ELCS | 2 (22.2%) | 0 (0%) | 0 (0%) | ||
| EMCS | 1 (11.1%) | 3 (30%) | 2 (20%) | ||
| Breastfeeding | 4 (44.4%) | 7 (70%) | 10 (100%) | 0.025 | C < B ~ A |
| Breastfeeding duration (months) | 9.7 ± 5.2 | 11.3 ± 6.2 | 10.8 ± 7.2 | 0.927 |
| Variable | Italian IQSEC2 Population (ID Patient #) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | |
| Gender | M | F | F | M | M | F | M | F | F | F | F | F | M | M | M | F | M | M | F |
| Regression followed by recovery or stabilization | − | − | − | + | + | − | + | − | + | − | − | − | − | − | − | − | − | − | − |
| Main criteria | |||||||||||||||||||
| Partial/complete loss of acquired purposeful hand skills | + | + | − | + | + | + | + | + | − | − | + | − | + | + | + | + | + | − | − |
| Partial or complete loss of acquired spoken language | + | + | − | + | + | + | + | + | + | + | + | + | + | + | − | + | + | + | + |
| Gait abnormalities | + | − | − | + | + | + | + | + | + | + | − | + | + | + | − | − | + | + | + |
| Stereotypic hand movements | + | − | + | + | + | + | + | + | + | + | + | − | + | + | − | + | + | + | − |
| Total criteria (n) | 4 | 2 | 1 | 4 | 4 | 4 | 4 | 3 | 3 | 3 | 3 | 2 | 4 | 4 | 1 | 3 | 4 | 3 | 2 |
| Exclusion criteria | |||||||||||||||||||
| Secondary brain injury | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Grossly abnormal PM development in first 6 months | − | − | − | + | + | − | + | − | + | + | + | − | + | + | + | + | + | + | − |
| Total exclusion criteria (n) | − | − | − | − | + | − | + | + | + | + | + | − | + | + | + | + | + | + | − |
| Supportive criteria | |||||||||||||||||||
| Breathing disturbances when awake | − | − | + | − | − | − | − | + | + | − | + | − | − | − | − | − | − | − | − |
| Bruxism when awake | + | − | − | + | − | + | + | − | − | + | + | + | − | − | + | + | + | + | + |
| Impaired sleep pattern | + | − | + | + | − | + | + | + | + | − | − | − | + | + | − | + | + | + | + |
| Abnormal muscle tone | + | − | − | + | + | + | − | + | − | − | − | + | + | + | − | + | + | + | − |
| Peripheral vasomotor disturbances | − | − | + | + | − | + | + | + | + | + | + | + | + | − | − | + | − | − | + |
| Scoliosis/kyphosis | + | − | + | + | − | − | − | + | − | − | − | + | + | − | − | + | + | − | − |
| Growth retardation | + | − | − | + | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Small cold hands and feet | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Inappropriate laughing/screaming spells | − | − | + | + | − | + | + | − | + | − | − | − | − | + | + | + | − | + | + |
| Diminished response to pain | + | − | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − | − |
| Eye pointing | − | − | + | − | + | + | + | − | − | + | + | − | − | + | + | − | + | + | + |
| Total supportive criteria (n) | 6 | − | 7 | 8 | 4 | 7 | 6 | 6 | 5 | 3 | 5 | 5 | 5 | 5 | 4 | 7 | 6 | 5 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leoncini, S.; Boasiako, L.; Lopergolo, D.; Altamura, M.; Fazzi, C.; Canitano, R.; Grosso, S.; Meloni, I.; Baldassarri, M.; Croci, S.; et al. Natural Course of IQSEC2-Related Encephalopathy: An Italian National Structured Survey. Children 2023, 10, 1442. https://doi.org/10.3390/children10091442
Leoncini S, Boasiako L, Lopergolo D, Altamura M, Fazzi C, Canitano R, Grosso S, Meloni I, Baldassarri M, Croci S, et al. Natural Course of IQSEC2-Related Encephalopathy: An Italian National Structured Survey. Children. 2023; 10(9):1442. https://doi.org/10.3390/children10091442
Chicago/Turabian StyleLeoncini, Silvia, Lidia Boasiako, Diego Lopergolo, Maria Altamura, Caterina Fazzi, Roberto Canitano, Salvatore Grosso, Ilaria Meloni, Margherita Baldassarri, Susanna Croci, and et al. 2023. "Natural Course of IQSEC2-Related Encephalopathy: An Italian National Structured Survey" Children 10, no. 9: 1442. https://doi.org/10.3390/children10091442