



Exploring the Genetic Landscape of Childhood Glaucoma



Abstract
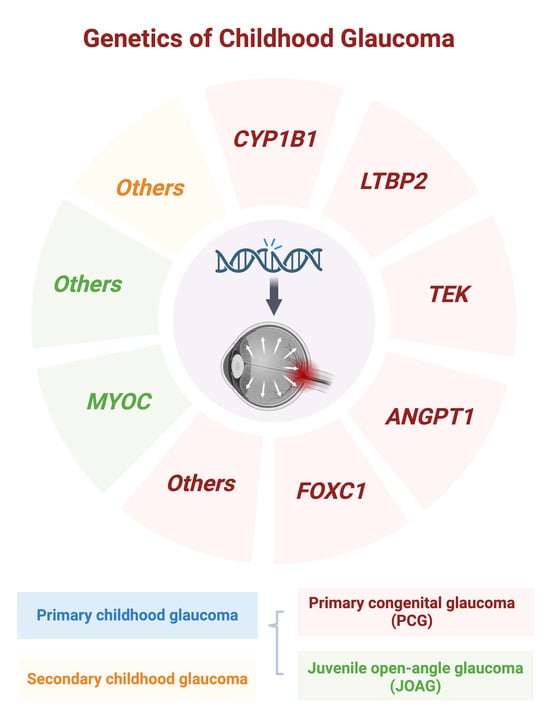
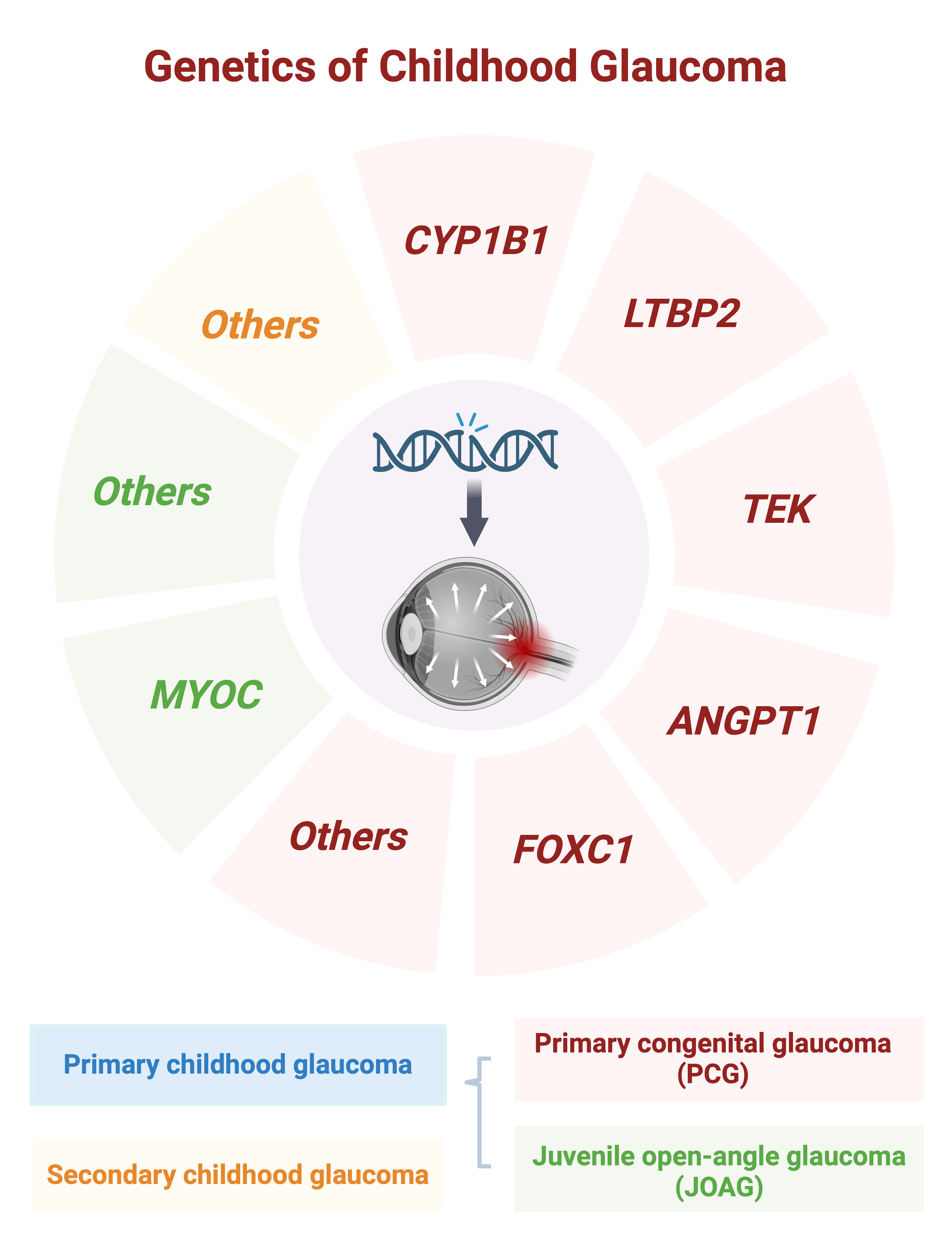
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Primary Congenital Glaucoma (PCG)
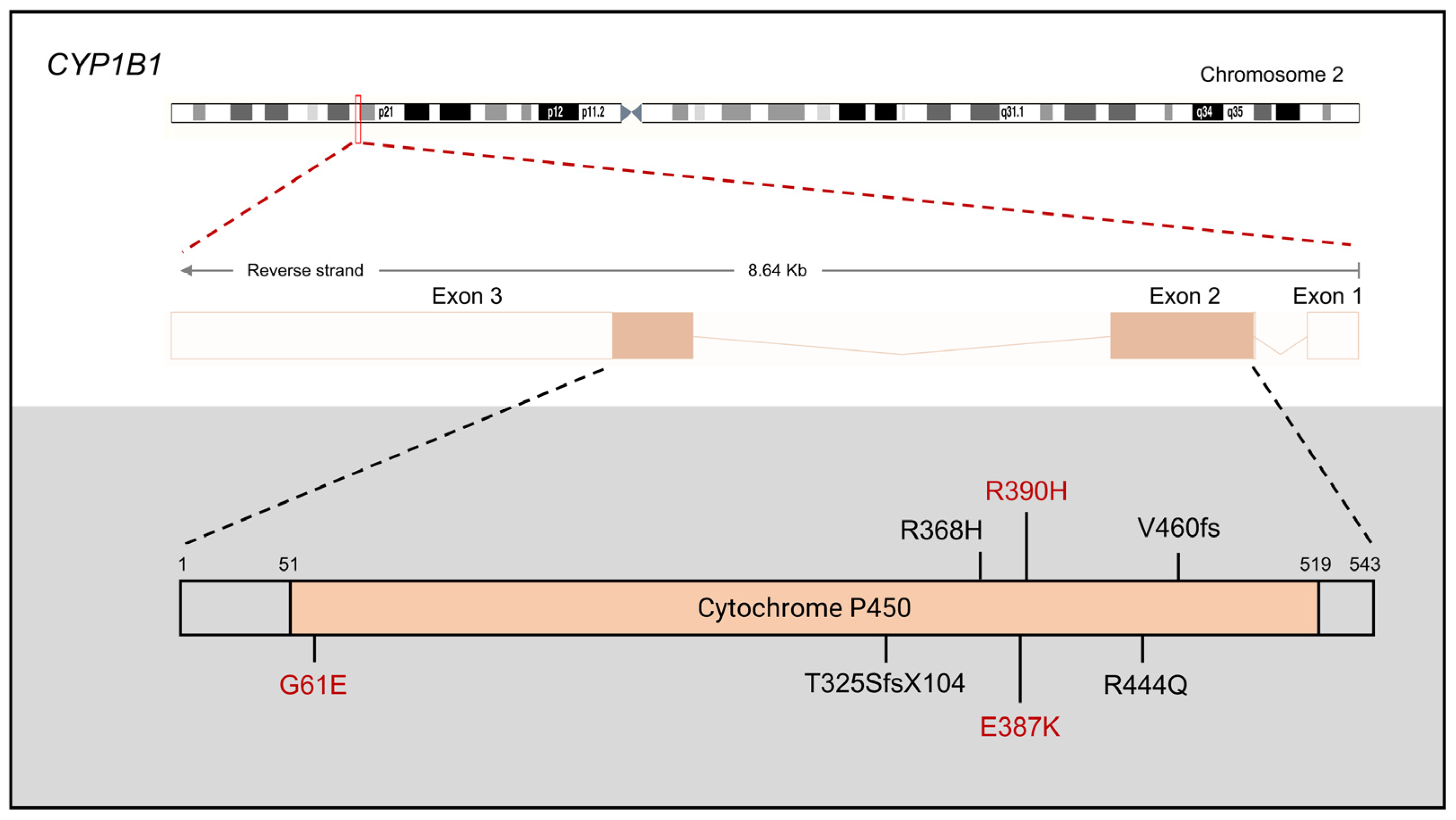
2.1. CYP1B1
2.1.1. CYP1B1 Mutations in PCG
2.1.2. CYP1B1 Protein Function in PCG
2.1.3. CYP1B1 Animal Models
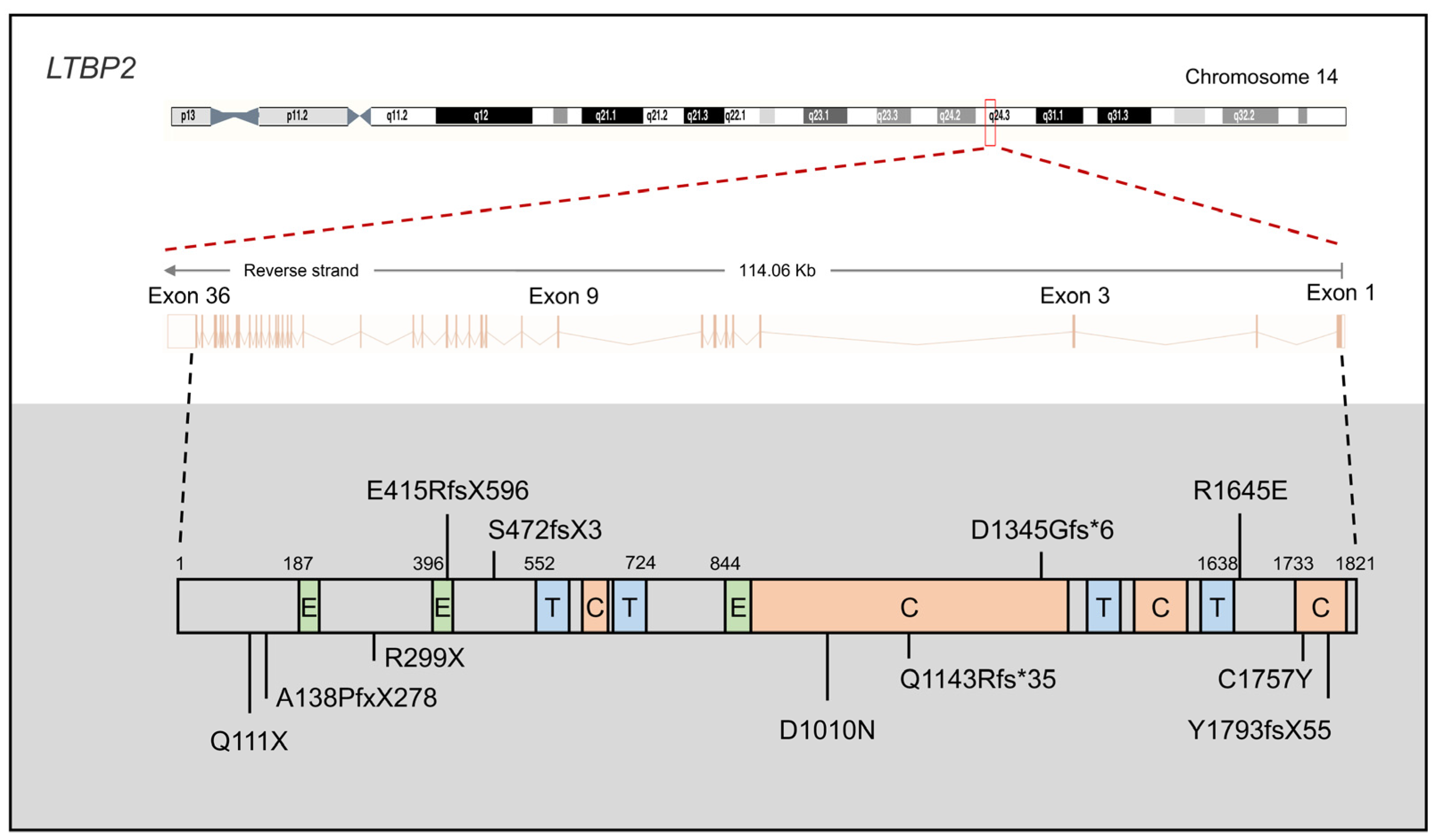
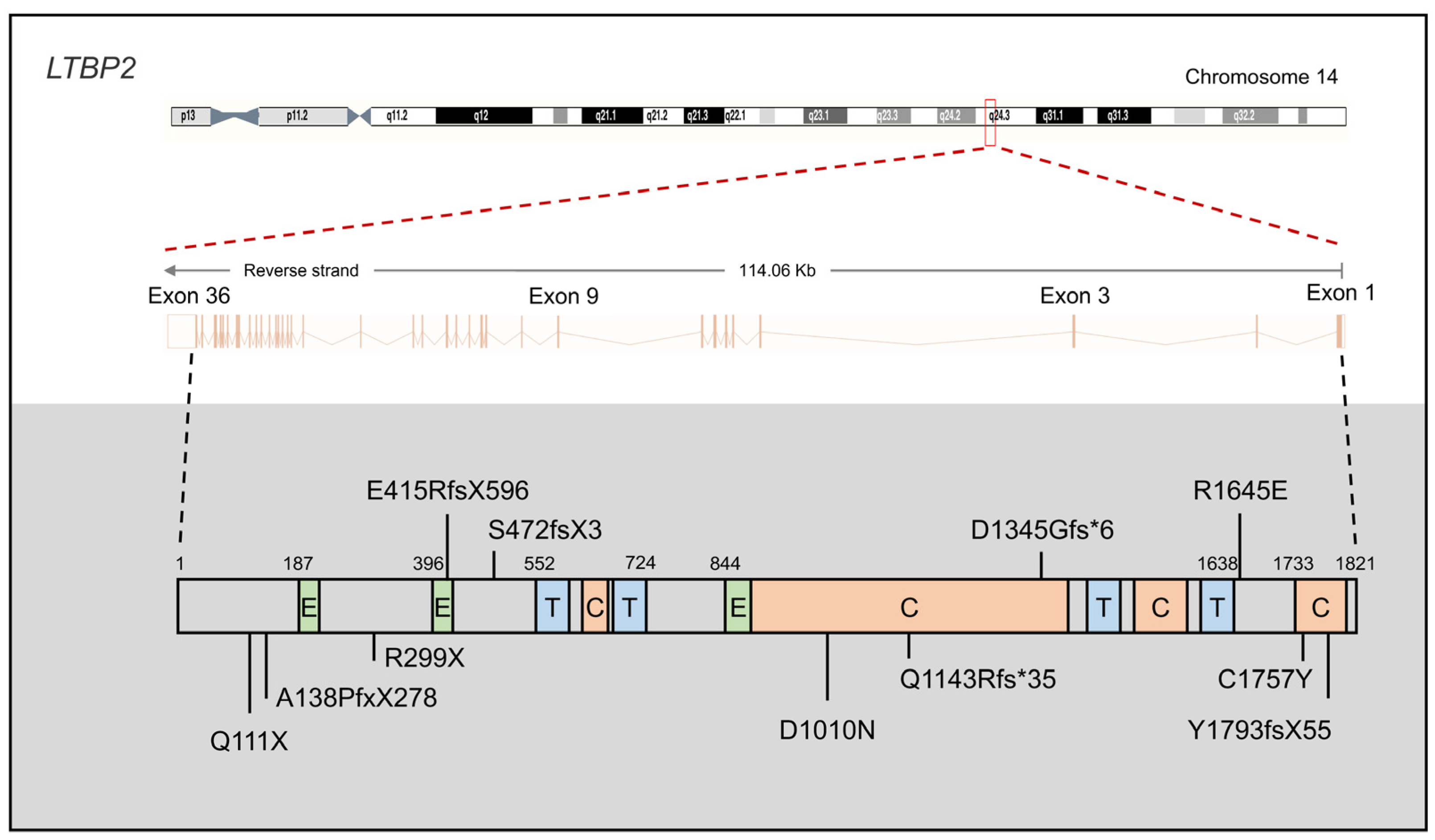
2.2. LTBP2
2.2.1. LTBP2 Mutations in PCG
2.2.2. LTBP2 Protein Function in PCG
2.2.3. LTBP2 Mouse Models
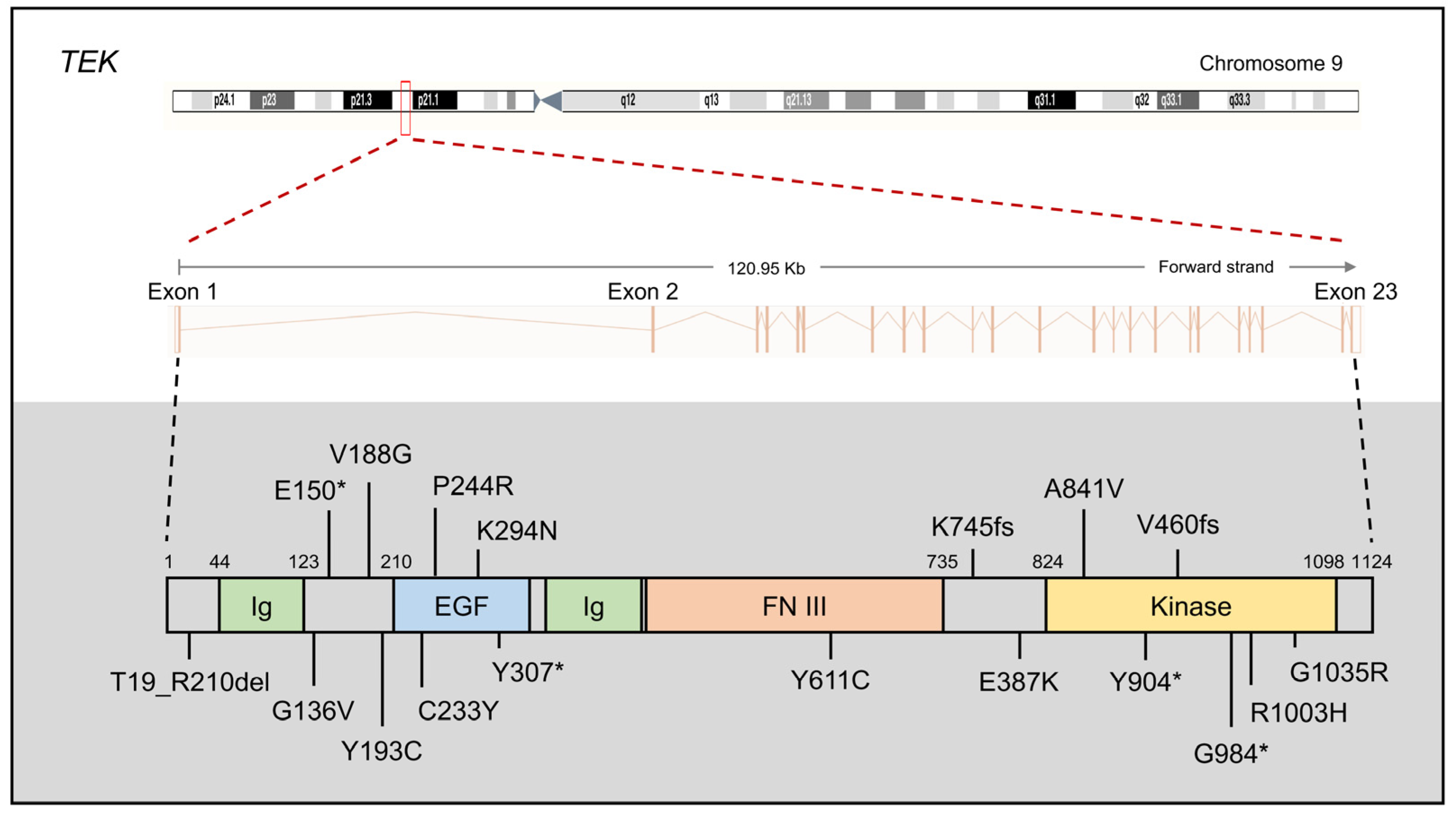
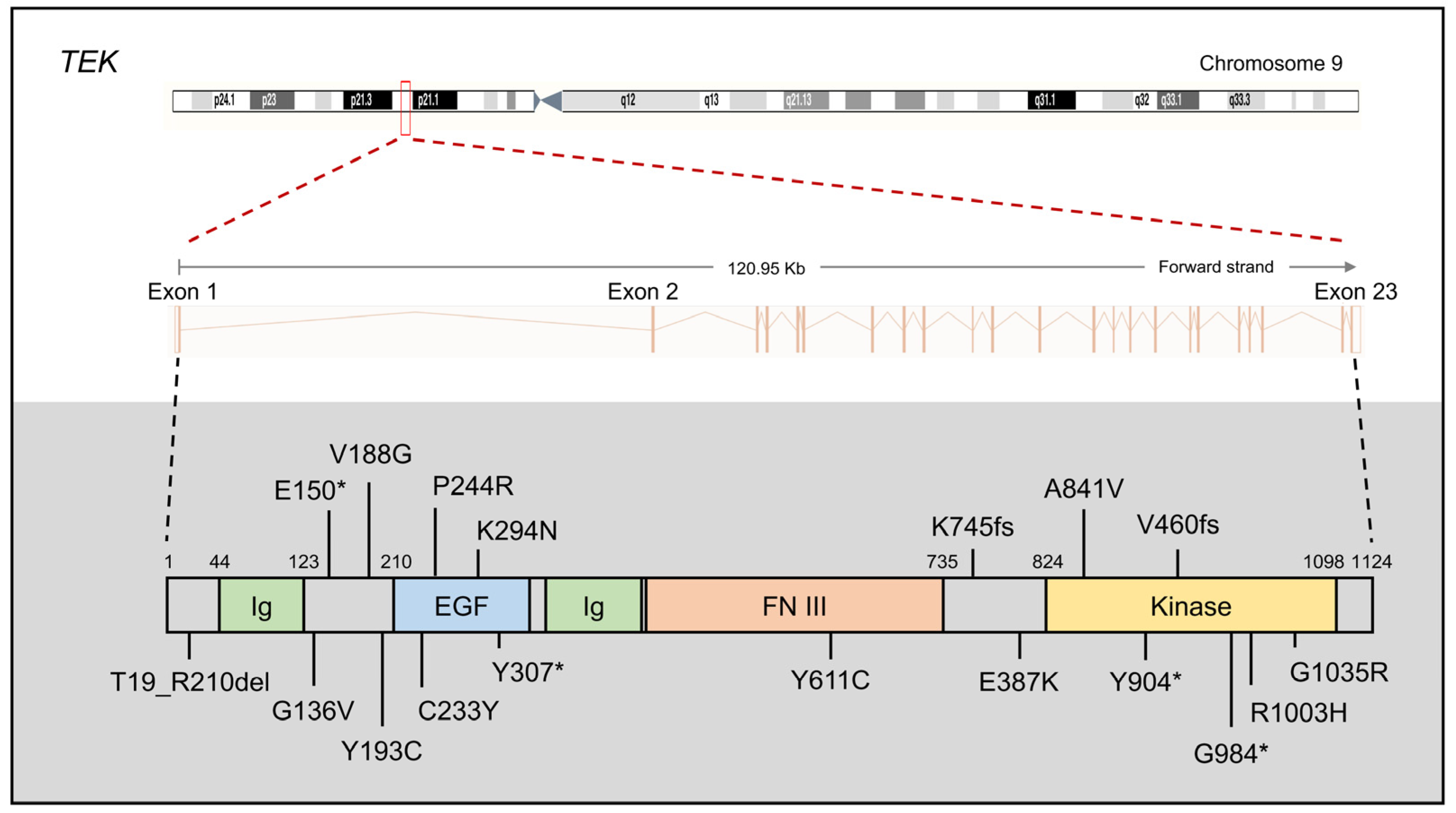
2.3. TEK and ANGPT1
2.3.1. TEK and ANGPT1 Mutations in PCG
2.3.2. The Protein Function of TEK and ANGPT1
2.3.3. TEK and ANGPT1 Mouse Models
2.4. FOXC1
3. Juvenile Open-Angle Glaucoma (JOAG)
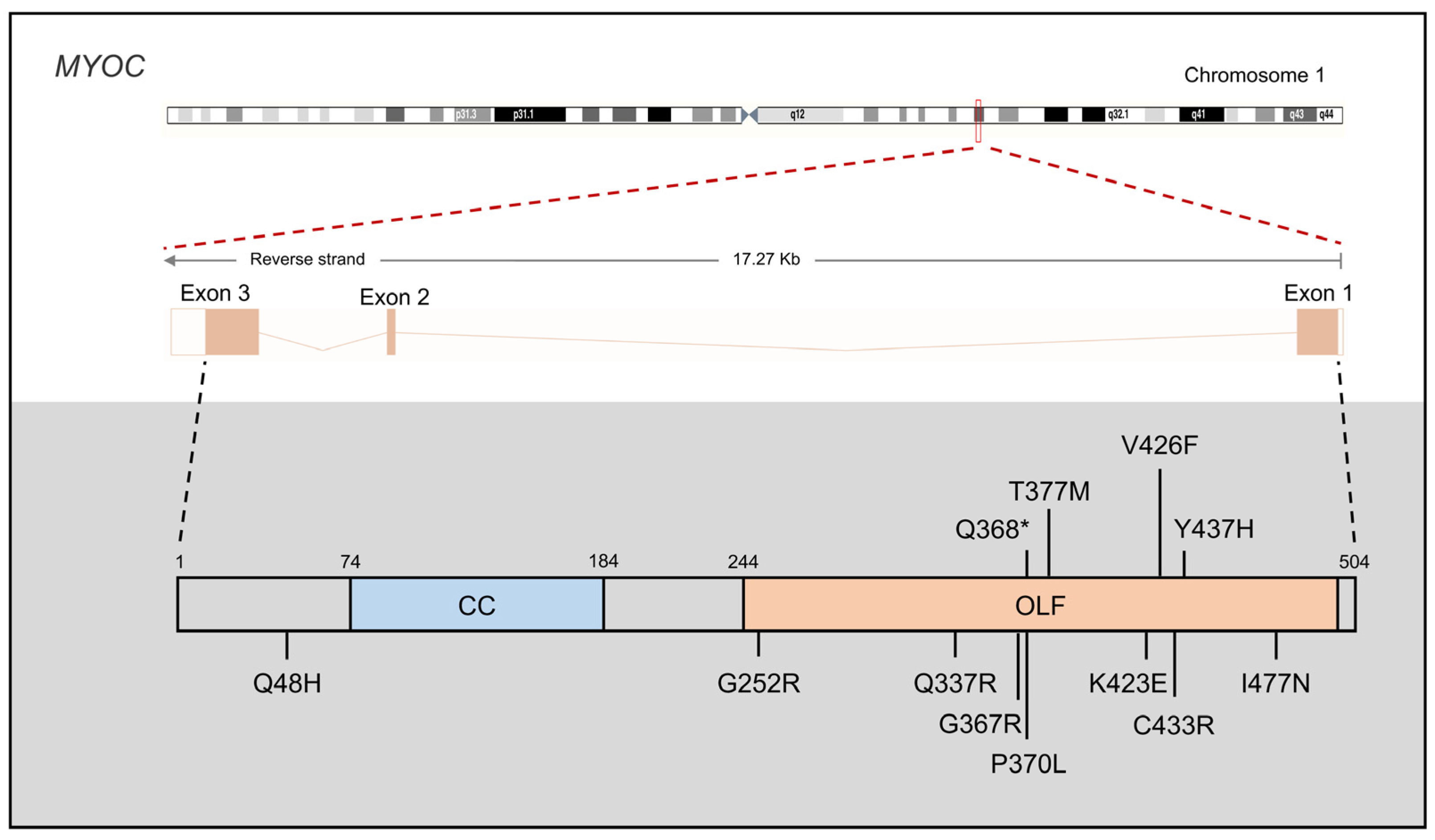
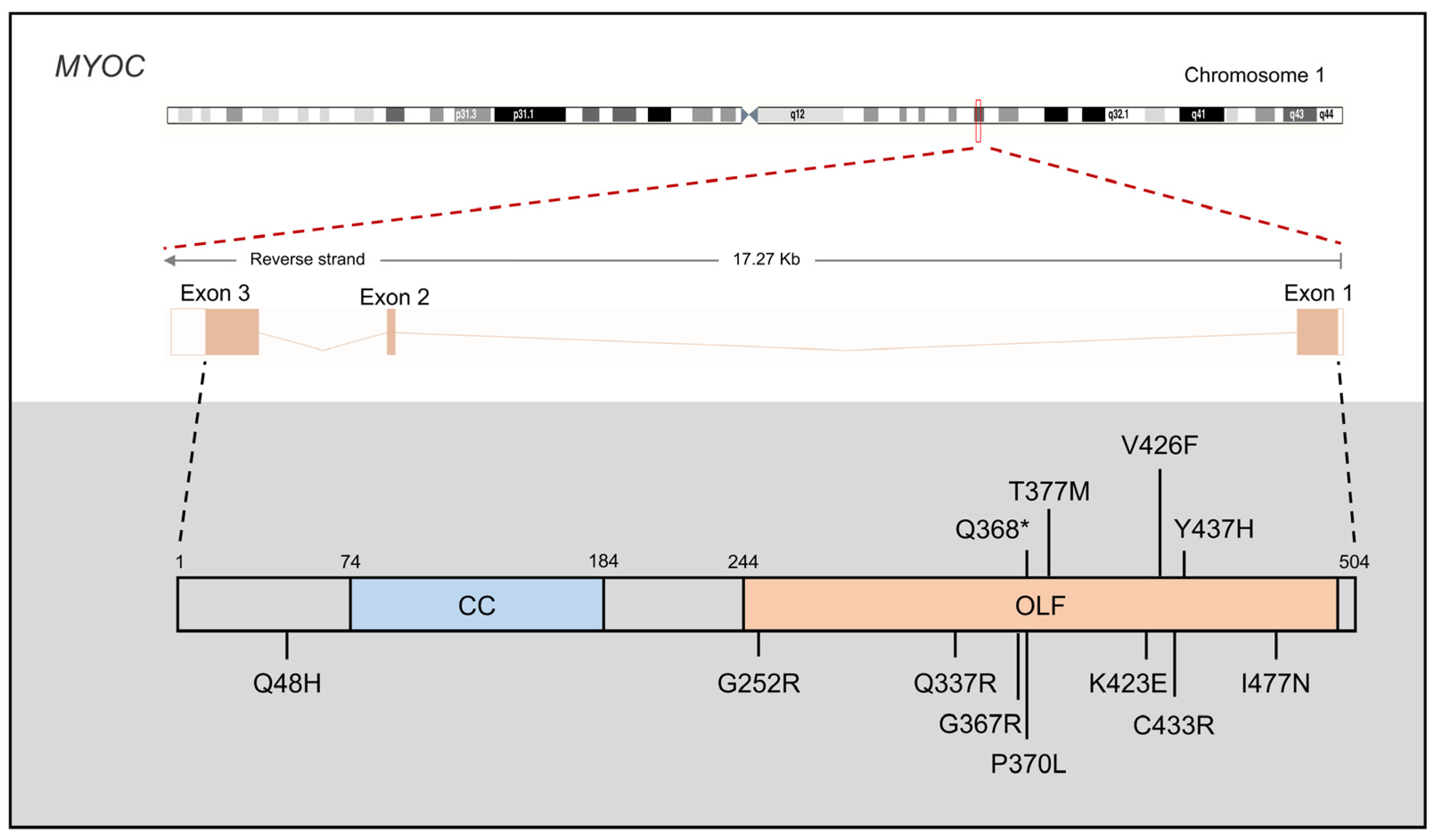
3.1. MYOC
3.1.1. MYOC Mutations in JOAG
3.1.2. MYOC Protein Function in PCG
3.1.3. MYOC Mouse Models
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Minegishi, Y.; Nakayama, M.; Iejima, D.; Kawase, K.; Iwata, T. Significance of Optineurin Mutations in Glaucoma and Other Diseases. Prog. Retin. Eye Res. 2016, 55, 149–181. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Pasquale, L.R. Genetics of Glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Iwata, T. Molecular Genetics of Inherited Normal Tension Glaucoma. Indian J. Ophthalmol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Knight, L.S.W.; Ruddle, J.B.; Taranath, D.A.; Goldberg, I.; Smith, J.E.H.; Gole, G.; Chiang, M.Y.; Willett, F.; D’Mellow, G.; Breen, J.; et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology 2021, 128, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; World Glaucoma Association (Eds.) Childhood Glaucoma: The 9th Consensus Report of the World Glaucoma Association; [...Vancouver, July, 17–20, 2013]; Consensus Series/World Glaucoma Association; Kugler: Amsterdam, The Netherlands, 2013; ISBN 978-90-6299-239-3. [Google Scholar]
- Karaconji, T.; Zagora, S.; Grigg, J.R. Approach to Childhood Glaucoma: A Review. Clin. Exp. Ophthalmol. 2022, 50, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Mokbel, T.H.; El Hefney, E.M.; Hagras, S.M.; ALNagdy, A.A.; Badawi, A.E.; Kasem, M.A.; El Shaer, S.M. Childhood Glaucoma Profile in Dakahelia, Egypt: A Retrospective Study. Int. J. Ophthalmol. 2018, 11, 674–680. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, Y.M.; Elhusseiny, A.M.; Gawdat, G.I.; Esmael, A.F.; Elhilali, H.M. Childhood Glaucoma Profile in a Tertiary Centre in Egypt According to the Childhood Glaucoma Research Network Classification. PLoS ONE 2023, 18, e0279874. [Google Scholar] [CrossRef]
- Hoguet, A.; Grajewski, A.; Hodapp, E.; Chang, T.P. A Retrospective Survey of Childhood Glaucoma Prevalence According to Childhood Glaucoma Research Network Classification. Indian J. Ophthalmol. 2016, 64, 118. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.L.; Gracitelli, C.P.B.; Rolim-de-Moura, C. Childhood Glaucoma Profile in a Brazilian Tertiary Care Center Using Childhood Glaucoma Research Network Classification. J. Glaucoma 2021, 30, 129–133. [Google Scholar] [CrossRef]
- Senthil, S.; Badakere, S.; Ganesh, J.; Krishnamurthy, R.; Dikshit, S.; Choudhari, N.; Garudadri, C.; Mandal, A. Profile of Childhood Glaucoma at a Tertiary Center in South India. Indian J. Ophthalmol. 2019, 67, 358. [Google Scholar] [CrossRef]
- Shen, R.; Li, V.S.W.; Wong, M.O.M.; Chan, P.P.M. Pediatric Glaucoma—From Screening, Early Detection to Management. Children 2023, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.J.; Hedberg-Buenz, A.; DeLuca, A.P.; Stone, E.M.; Alward, W.L.M.; Fingert, J.H. Primary Congenital and Developmental Glaucomas. Hum. Mol. Genet. 2017, 26, R28–R36. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Tompson, S.W.; Thomson, B.R.; Siggs, O.M.; Kizhatil, K.; Yamaguchi, S.; Feng, L.; Limviphuvadh, V.; Whisenhunt, K.N.; Maurer-Stroh, S.; et al. Angiopoietin Receptor TEK Mutations Underlie Primary Congenital Glaucoma with Variable Expressivity. J. Clin. Investig. 2016, 126, 2575–2587. [Google Scholar] [CrossRef]
- Lim, S.-H.; Tran-Viet, K.-N.; Yanovitch, T.L.; Freedman, S.F.; Klemm, T.; Call, W.; Powell, C.; Ravichandran, A.; Metlapally, R.; Nading, E.B.; et al. CYP1B1, MYOC, and LTBP2 Mutations in Primary Congenital Glaucoma Patients in the United States. Am. J. Ophthalmol. 2013, 155, 508–517.e5. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.J.; Wiggs, J.L. Glaucoma: Genes, Phenotypes, and New Directions for Therapy. J. Clin. Investig. 2010, 120, 3064–3072. [Google Scholar] [CrossRef]
- Sarfarazi, M.; Akarsu, N.A.; Hossain, A.; Turacli, E.M.; Aktan, G.S.; Barsoum-Homsy, M.; Chevrette, L.; Sayli, S.B. Assignment of a Locus (GLC3A) for Primary Congenital Glaucoma (Buphthalmos) to 2p21 and Evidence for Genetic Heterogeneity. Genomics 1995, 30, 171–177. [Google Scholar] [CrossRef]
- Akarsu, A. A Second Locus (GLC3B) for Primary Congenital Glaucoma (Buphthalmos) Maps to the 1p36 Region. Hum. Mol. Genet. 1996, 5, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, I.; Sarfarazi, M. The Third Genetic Locus (GLC3C) for Primary Congenital Glaucoma (PCG) Maps to Chromosome 14q24.3. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3015. [Google Scholar]
- Firasat, S.; Riazuddin, S.A.; Hejtmancik, J.F.; Riazuddin, S. Primary Congenital Glaucoma Localizes to Chromosome 14q24.2-24.3 in Two Consanguineous Pakistani Families. Mol. Vis. 2008, 14, 1659–1665. [Google Scholar]
- Shue, A.; Wong, M.O.; Freedman, S.F. Primary Congenital Glaucoma. In Albert and Jakobiec’s Principles and Practice of Ophthalmology; Albert, D., Miller, J., Azar, D., Young, L.H., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–40. ISBN 978-3-319-90495-5. [Google Scholar]
- Stoilov, I. Identification of Three Different Truncating Mutations in Cytochrome P4501B1 (CYP1B1) as the Principal Cause of Primary Congenital Glaucoma (Buphthalmos) in Families Linked to the GLC3A Locus on Chromosome 2p21. Hum. Mol. Genet. 1997, 6, 641–647. [Google Scholar] [CrossRef]
- Ali, M.; McKibbin, M.; Booth, A.; Parry, D.A.; Jain, P.; Riazuddin, S.A.; Hejtmancik, J.F.; Khan, S.N.; Firasat, S.; Shires, M.; et al. Null Mutations in LTBP2 Cause Primary Congenital Glaucoma. Am. J. Hum. Genet. 2009, 84, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Narooie-Nejad, M.; Paylakhi, S.H.; Shojaee, S.; Fazlali, Z.; Rezaei Kanavi, M.; Nilforushan, N.; Yazdani, S.; Babrzadeh, F.; Suri, F.; Ronaghi, M.; et al. Loss of Function Mutations in the Gene Encoding Latent Transforming Growth Factor Beta Binding Protein 2, LTBP2, Cause Primary Congenital Glaucoma. Hum. Mol. Genet. 2009, 18, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Kaur, K.; Rao, K.N.; Mandal, A.K.; Kaur, I.; Parikh, R.S.; Thomas, R. The Transcription Factor Gene FOXC1 Exhibits a Limited Role in Primary Congenital Glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 75. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.R.; Souma, T.; Tompson, S.W.; Onay, T.; Kizhatil, K.; Siggs, O.M.; Feng, L.; Whisenhunt, K.N.; Yanovitch, T.L.; Kalaydjieva, L.; et al. Angiopoietin-1 Is Required for Schlemm’s Canal Development in Mice and Humans. J. Clin. Investig. 2017, 127, 4421–4436. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.M.; Wo, Y.-Y.P.; Stewart, J.; Hawkins, A.L.; Griffin, C.A.; Sutter, T.R.; Greenlee, W.F. Isolation and Characterization of the Human Cytochrome P450 CYP1B1 Gene. J. Biol. Chem. 1996, 271, 28324–28330. [Google Scholar] [CrossRef]
- Bejjani, B.A. Multiple CYP1B1 Mutations and Incomplete Penetrance in an Inbred Population Segregating Primary Congenital Glaucoma Suggest Frequent de Novo Events and a Dominant Modifier Locus. Hum. Mol. Genet. 2000, 9, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Chitsazian, F.; Tusi, B.K.; Elahi, E.; Saroei, H.A.; Sanati, M.H.; Yazdani, S.; Pakravan, M.; Nilforooshan, N.; Eslami, Y.; Mehrjerdi, M.A.Z.; et al. CYP1B1 Mutation Profile of Iranian Primary Congenital Glaucoma Patients and Associated Haplotypes. J. Mol. Diagn. 2007, 9, 382–393. [Google Scholar] [CrossRef]
- Stoilov, I.R.; Costa, V.P.; Vasconcellos, J.P.C.; Melo, M.B.; Betinjane, A.J.; Carani, J.C.E.; Oltrogge, E.V.; Sarfarazi, M. Molecular Genetics of Primary Congenital Glaucoma in Brazil. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1820–1827. [Google Scholar]
- Chakrabarti, S.; Kaur, K.; Mandal, A. Primary Congenital Glaucoma and the Involvement of CYP1B1. Middle East. Afr. J. Ophthalmol. 2011, 18, 7. [Google Scholar] [CrossRef]
- Cascella, R.; Strafella, C.; Germani, C.; Novelli, G.; Ricci, F.; Zampatti, S.; Giardina, E. The Genetics and the Genomics of Primary Congenital Glaucoma. BioMed Res. Int. 2015, 2015, 321291. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Al Ghanim, K.; Yavarna, T.; El Akoum, M.; Samara, M.; Chandra, P.; Al-Dewik, N. Effects of Consanguinity in a Cohort of Subjects with Certain Genetic Disorders in Qatar. Mol. Genet. Genom. Med. 2020, 8, e1051. [Google Scholar] [CrossRef]
- Jalal Abbasi-Shavazi, M.; McDonald, P.; Hosseini-Chavoshi, M. Modernization or Cultural Maintenance: The Practice of Consanguineous Marriage in Iran. J. Biosoc. Sci. 2008, 40, 911–933. [Google Scholar] [CrossRef]
- Bittles, A.H.; Black, M.L. Consanguinity, Human Evolution, and Complex Diseases. Proc. Natl. Acad. Sci. USA 2010, 107, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Bouhenni, R.; Benmerzouga, I. Geographical Variability in CYP1B1 Mutations in Primary Congenital Glaucoma. JCM 2022, 11, 2048. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Liu, G.; Liu, W.; Deng, Y.; Zheng, L.; Zhang, S.; Feng, G. Bioinformatics Analysis of CYP1B1 Mutation Hotspots in Chinese Primary Congenital Glaucoma Patients. Biosci. Rep. 2018, 38, BSR20180056. [Google Scholar] [CrossRef]
- Mashima, Y.; Suzuki, Y.; Sergeev, Y.; Ohtake, Y.; Tanino, T.; Kimura, I.; Miyata, H.; Aihara, M.; Tanihara, H.; Inatani, M.; et al. Novel Cytochrome P4501B1 (CYP1B1) Gene Mutations in Japanese Patients with Primary Congenital Glaucoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2211–2216. [Google Scholar]
- Kim, H.-J.; Suh, W.; Park, S.C.; Kim, C.Y.; Park, K.H.; Kook, M.S.; Kim, Y.Y.; Kim, C.-S.; Park, C.K.; Ki, C.-S.; et al. Mutation Spectrum of CYP1B1 and MYOC Genes in Korean Patients with Primary Congenital Glaucoma. Mol. Vis. 2011, 17, 2093–2101. [Google Scholar]
- De Melo, M.B.; Mandal, A.K.; Tavares, I.M.; Ali, M.H.; Kabra, M.; De Vasconcellos, J.P.C.; Senthil, S.; Sallum, J.M.F.; Kaur, I.; Betinjane, A.J.; et al. Genotype-Phenotype Correlations in CYP1B1-Associated Primary Congenital Glaucoma Patients Representing Two Large Cohorts from India and Brazil. PLoS ONE 2015, 10, e0127147. [Google Scholar] [CrossRef] [PubMed]
- López-Garrido, M.-P.; Medina-Trillo, C.; Morales-Fernandez, L.; Garcia-Feijoo, J.; Martínez-de-la-Casa, J.-M.; García-Antón, M.; Escribano, J. Null CYP1B1 Genotypes in Primary Congenital and Nondominant Juvenile Glaucoma. Ophthalmology 2013, 120, 716–723. [Google Scholar] [CrossRef]
- Li, F.; Zhu, W.; Gonzalez, F.J. Potential Role of CYP1B1 in the Development and Treatment of Metabolic Diseases. Pharmacol. Ther. 2017, 178, 18–30. [Google Scholar] [CrossRef]
- Choudhary, D.; Jansson, I.; Sarfarazi, M.; Schenkman, J.B. Physiological Significance and Expression of P450s in the Developing Eye. Drug Metab. Rev. 2006, 38, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Doshi, M.; Marcus, C.; Bejjani, B.A.; Edward, D.P. Immunolocalization of CYP1B1 in Normal, Human, Fetal and Adult Eyes. Exp. Eye Res. 2006, 82, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, S.; Sorenson, C.M.; Teixeira, L.; Dubielzig, R.R.; Peters, D.M.; Conway, S.J.; Jefcoate, C.R.; Sheibani, N. Cyp1b1 Mediates Periostin Regulation of Trabecular Meshwork Development by Suppression of Oxidative Stress. Mol. Cell. Biol. 2013, 33, 4225–4240. [Google Scholar] [CrossRef] [PubMed]
- Falero-Perez, J.; Song, Y.-S.; Sorenson, C.M.; Sheibani, N. CYP1B1: A Key Regulator of Redox Homeostasis. Trends Cell Mol. Biol. 2018, 13, 27–45. [Google Scholar]
- Palenski, T.L.; Sorenson, C.M.; Jefcoate, C.R.; Sheibani, N. Lack of Cyp1b1 Promotes the Proliferative and Migratory Phenotype of Perivascular Supporting Cells. Lab. Investig. 2013, 93, 646–662. [Google Scholar] [CrossRef]
- Tang, Y.; Scheef, E.A.; Wang, S.; Sorenson, C.M.; Marcus, C.B.; Jefcoate, C.R.; Sheibani, N. CYP1B1 Expression Promotes the Proangiogenic Phenotype of Endothelium through Decreased Intracellular Oxidative Stress and Thrombospondin-2 Expression. Blood 2009, 113, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.R.; Ramsden, D.B.; Williams, A.C. Cytochrome P450 1B1 mRNA in the Human Central Nervous System. Mol. Pathol. 1998, 51, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, D.; Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Metabolism of Retinoids and Arachidonic Acid by Human and Mouse Cytochrome P450 1b1. Drug Metab. Dispos. 2004, 32, 840–847. [Google Scholar] [CrossRef]
- Korkmaz, A.; Topal, T.; Tan, D.-X.; Reiter, R.J. Role of Melatonin in Metabolic Regulation. Rev. Endocr. Metab. Disord. 2009, 10, 261–270. [Google Scholar] [CrossRef]
- Alkozi, H.A.; Navarro, G.; Franco, R.; Pintor, J. Melatonin and the Control of Intraocular Pressure. Prog. Retin. Eye Res. 2020, 75, 100798. [Google Scholar] [CrossRef]
- Chang, T.K.H.; Chen, J.; Yang, G.; Yeung, E.Y.H. Inhibition of Procarcinogen-bioactivating Human CYP1A1, CYP1A2 and CYP1B1 Enzymes by Melatonin. J. Pineal Res. 2010, 48, 55–64. [Google Scholar] [CrossRef]
- Malik, K.U.; Jennings, B.L.; Yaghini, F.A.; Sahan-Firat, S.; Song, C.Y.; Estes, A.M.; Fang, X.R. Contribution of Cytochrome P450 1B1 to Hypertension and Associated Pathophysiology: A Novel Target for Antihypertensive Agents. Prostaglandins Other Lipid Mediat. 2012, 98, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Masferrer, J.L.; Dunn, M.W.; Schwartzman, M.L. 12(R)-Hydroxyeicosatetraenoic Acid, an Endogenous Corneal Arachidonate Metabolite, Lowers Intraocular Pressure in Rabbits. Investig. Ophthalmol. Vis. Sci. 1990, 31, 535–539. [Google Scholar]
- Aribindi, K.; Guerra, Y.; Lee, R.K.; Bhattacharya, S.K. Comparative Phospholipid Profiles of Control and Glaucomatous Human Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3037. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, I.; Rezaie, T.; Jansson, I.; Schenkman, J.B.; Sarfarazi, M. Expression of Cytochrome P4501b1 (Cyp1b1) during Early Murine Development. Mol. Vis. 2004, 10, 629–636. [Google Scholar] [PubMed]
- Sheibani, N.; Zhao, Y.; Sorenson, C. Cytochrome P450 1B1 and Primary Congenital Glaucoma. J. Ophthalmic Vis. Res. 2015, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Buters, J.T.M.; Sakai, S.; Richter, T.; Pineau, T.; Alexander, D.L.; Savas, U.; Doehmer, J.; Ward, J.M.; Jefcoate, C.R.; Gonzalez, F.J. Cytochrome P450 CYP1B1 Determines Susceptibility to 7,12-Dimethylbenz[a]Anthracene-Induced Lymphomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Libby, R.T.; Smith, R.S.; Savinova, O.V.; Zabaleta, A.; Martin, J.E.; Gonzalez, F.J.; John, S.W.M. Modification of Ocular Defects in Mouse Developmental Glaucoma Models by Tyrosinase. Science 2003, 299, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.B.C.; Zhao, Y.; Dubielzig, R.R.; Sorenson, C.M.; Sheibani, N. Ultrastructural Abnormalities of the Trabecular Meshwork Extracellular Matrix in Cyp1b1-Deficient Mice. Vet. Pathol. 2015, 52, 397–403. [Google Scholar] [CrossRef]
- Amirmokhtari, N.; Foresi, B.D.; Dewan, S.S.; Bouhenni, R.A.; Smith, M.A. Absence of Cytochrome P450-1b1 Increases Susceptibility of Pressure-Induced Axonopathy in the Murine Retinal Projection. Front. Cell Dev. Biol. 2021, 9, 636321. [Google Scholar] [CrossRef]
- Williams, A.L.; Eason, J.; Chawla, B.; Bohnsack, B.L. Cyp1b1 Regulates Ocular Fissure Closure Through a Retinoic Acid–Independent Pathway. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1084. [Google Scholar] [CrossRef] [PubMed]
- Azmanov, D.N.; Dimitrova, S.; Florez, L.; Cherninkova, S.; Draganov, D.; Morar, B.; Saat, R.; Juan, M.; Arostegui, J.I.; Ganguly, S.; et al. LTBP2 and CYP1B1 Mutations and Associated Ocular Phenotypes in the Roma/Gypsy Founder Population. Eur. J. Hum. Genet. 2011, 19, 326–333. [Google Scholar] [CrossRef]
- Micheal, S.; Siddiqui, S.N.; Zafar, S.N.; Iqbal, A.; Khan, M.I.; Den Hollander, A.I. Identification of Novel Variants in LTBP2 and PXDN Using Whole-Exome Sequencing in Developmental and Congenital Glaucoma. PLoS ONE 2016, 11, e0159259. [Google Scholar] [CrossRef] [PubMed]
- Rauf, B.; Irum, B.; Khan, S.Y.; Kabir, F.; Naeem, M.A.; Riazuddin, S.; Ayyagari, R.; Riazuddin, S.A. Novel Mutations in LTBP2 Identified in Familial Cases of Primary Congenital Glaucoma. Mol. Vis. 2020, 26, 14–25. [Google Scholar] [PubMed]
- Saharinen, J.; Hyytiäinen, M.; Taipale, J.; Keski-Oja, J. Latent Transforming Growth Factor-β Binding Proteins (LTBPs)—Structural Extracellular Matrix Proteins for Targeting TGF-β Action. Cytokine Growth Factor Rev. 1999, 10, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-Binding Proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hirani, R.; Hanssen, E.; Gibson, M.A. LTBP-2 Specifically Interacts with the Amino-Terminal Region of Fibrillin-1 and Competes with LTBP-1 for Binding to This Microfibrillar Protein. Matrix Biol. 2007, 26, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Suga, A.; Kimura, I.; Kimura, C.; Minegishi, Y.; Nakayama, M.; Yoshitake, K.; Iejima, D.; Minematsu, N.; Yamamoto, M.; et al. METTL23 Mutation Alters Histone H3R17 Methylation in Normal-Tension Glaucoma. J. Clin. Investig. 2022, 132, e153589. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.N.; Autar, R. Flow of Aqueous Humor in the Canal of Schlemm. Math. Biosci. 1989, 93, 53–78. [Google Scholar] [CrossRef]
- Carreon, T.; Van Der Merwe, E.; Fellman, R.L.; Johnstone, M.; Bhattacharya, S.K. Aqueous Outflow—A Continuum from Trabecular Meshwork to Episcleral Veins. Prog. Retin. Eye Res. 2017, 57, 108–133. [Google Scholar] [CrossRef]
- Mahmud, N.; Eisner, C.; Purushothaman, S.; Storer, M.A.; Kaplan, D.R.; Miller, F.D. Nail-Associated Mesenchymal Cells Contribute to and Are Essential for Dorsal Digit Tip Regeneration. Cell Rep. 2022, 41, 111853. [Google Scholar] [CrossRef] [PubMed]
- Désir, J.; Sznajer, Y.; Depasse, F.; Roulez, F.; Schrooyen, M.; Meire, F.; Abramowicz, M. LTBP2 Null Mutations in an Autosomal Recessive Ocular Syndrome with Megalocornea, Spherophakia, and Secondary Glaucoma. Eur. J. Hum. Genet. 2010, 18, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Ohbayashi, T.; Fujikawa, Y.; Yoshida, H.; Akama, T.O.; Noda, K.; Horiguchi, M.; Kameyama, K.; Hata, Y.; Takahashi, K.; et al. Latent TGF-β Binding Protein-2 Is Essential for the Development of Ciliary Zonule Microfibrils. Hum. Mol. Genet. 2014, 23, 5672–5682. [Google Scholar] [CrossRef] [PubMed]
- Shipley, J.M.; Mecham, R.P.; Maus, E.; Bonadio, J.; Rosenbloom, J.; McCarthy, R.T.; Baumann, M.L.; Frankfater, C.; Segade, F.; Shapiro, S.D. Developmental Expression of Latent Transforming Growth Factor β Binding Protein 2 and Its Requirement Early in Mouse Development. Mol. Cell. Biol. 2000, 20, 4879–4887. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic Targeting of the Angiopoietin–TIE Pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Kizhatil, K.; Ryan, M.; Marchant, J.K.; Henrich, S.; John, S.W.M. Schlemm’s Canal Is a Unique Vessel with a Combination of Blood Vascular and Lymphatic Phenotypes That Forms by a Novel Developmental Process. PLoS Biol. 2014, 12, e1001912. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.R.; Heinen, S.; Jeansson, M.; Ghosh, A.K.; Fatima, A.; Sung, H.-K.; Onay, T.; Chen, H.; Yamaguchi, S.; Economides, A.N.; et al. A Lymphatic Defect Causes Ocular Hypertension and Glaucoma in Mice. J. Clin. Investig. 2014, 124, 4320–4324. [Google Scholar] [CrossRef] [PubMed]
- Young, T.L.; Whisenhunt, K.N.; Jin, J.; LaMartina, S.M.; Martin, S.M.; Souma, T.; Limviphuvadh, V.; Suri, F.; Souzeau, E.; Zhang, X.; et al. SVEP1 as a Genetic Modifier of TEK-Related Primary Congenital Glaucoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 6. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, Y.; Tan, C.; Sun, X.; Chen, X.; Chen, J. Screening and Functional Analysis of TEK Mutations in Chinese Children With Primary Congenital Glaucoma. Front. Genet. 2021, 12, 764509. [Google Scholar] [CrossRef]
- Choquet, H.; Paylakhi, S.; Kneeland, S.C.; Thai, K.K.; Hoffmann, T.J.; Yin, J.; Kvale, M.N.; Banda, Y.; Tolman, N.G.; Williams, P.A.; et al. A Multiethnic Genome-Wide Association Study of Primary Open-Angle Glaucoma Identifies Novel Risk Loci. Nat. Commun. 2018, 9, 2278. [Google Scholar] [CrossRef]
- Van Zyl, T.; Yan, W.; McAdams, A.; Peng, Y.-R.; Shekhar, K.; Regev, A.; Juric, D.; Sanes, J.R. Cell Atlas of Aqueous Humor Outflow Pathways in Eyes of Humans and Four Model Species Provides Insight into Glaucoma Pathogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10339–10349. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.Y. Orchestral Actions of Angiopoietin-1 in Vascular Regeneration. Trends Mol. Med. 2013, 19, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Toris, C.B.; Yablonski, M.E.; Wang, Y.-L.; Camras, C.B. Aqueous Humor Dynamics in the Aging Human Eye. Am. J. Ophthalmol. 1999, 127, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Alm, A.; Nilsson, S.F.E. Uveoscleral Outflow—A Review. Exp. Eye Res. 2009, 88, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Fu, Y.; Baird, P.N.; Guymer, R.H.; Das, T.; Iwata, T. Exploring the Contribution of ARMS2 and HTRA1 Genetic Risk Factors in Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2023, 97, 101159. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Kaestner, K.H. Fox Transcription Factors: From Development to Disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Dubey, S.; Choudhary, S.; Ratna, R.; Pandav, S.; Khan, A. Anterior Segment Dysgenesis: Insights into the Genetics and Pathogenesis. Indian J. Ophthalmol. 2022, 70, 2293. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Souzeau, E.; Pasutto, F.; Dubowsky, A.; Smith, J.E.H.; Taranath, D.; Pater, J.; Rait, J.L.; Narita, A.; Mauri, L.; et al. Prevalence of FOXC1 Variants in Individuals With a Suspected Diagnosis of Primary Congenital Glaucoma. JAMA Ophthalmol. 2019, 137, 348. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S. Haploinsufficiency of the Transcription Factors FOXC1 and FOXC2 Results in Aberrant Ocular Development. Hum. Mol. Genet. 2000, 9, 1021–1032. [Google Scholar] [CrossRef]
- Umali, J.; Hawkey-Noble, A.; French, C.R. Loss of Foxc1 in Zebrafish Reduces Optic Nerve Size and Cell Number in the Retinal Ganglion Cell Layer. Vis. Res. 2019, 156, 66–72. [Google Scholar] [CrossRef]
- Sheffield, V.C.; Stone, E.M.; Alward, W.L.M.; Drack, A.V.; Johnson, A.T.; Streb, L.M.; Nichols, B.E. Genetic Linkage of Familial Open Angle Glaucoma to Chromosome 1q21–Q31. Nat. Genet. 1993, 4, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Lynch, S.; Ynagi, G.; Maselli, M.; Auguste, J.; Del Bono, E.A.; Olson, L.M.; Haines, J.L. A Genomewide Scan Identifies Novel Early-Onset Primary Open-Angle Glaucoma Loci on 9q22 and 20p12. Am. J. Hum. Genet. 2004, 74, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.P.; Fan, B.J.; Canlas, O.; Wang, D.Y.; Dubois, S.; Tam, P.O.S.; Lam, D.S.C.; Raymond, V.; Ritch, R. A Genome-Wide Scan Maps a Novel Juvenile-Onset Primary Open Angle Glaucoma Locus to Chromosome 5q. Mol. Vis. 2006, 12, 85–92. [Google Scholar] [PubMed]
- Wang, D.Y.; Fan, B.J.; Chua, J.K.H.; Tam, P.O.S.; Leung, C.K.S.; Lam, D.S.C.; Pang, C.P. A Genome-Wide Scan Maps a Novel Juvenile-Onset Primary Open-Angle Glaucoma Locus to 15q. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5315. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Fingert, J.H.; Alward, W.L.M.; Nguyen, T.D.; Polansky, J.R.; Sunden, S.L.F.; Nishimura, D.; Clark, A.F.; Nystuen, A.; Nichols, B.E.; et al. Identification of a Gene That Causes Primary Open Angle Glaucoma. Science 1997, 275, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, M.; Zhang, Z.; Xue, H.; Chen, X.; Ji, Y. Physiological Function of Myocilin and Its Role in the Pathogenesis of Glaucoma in the Trabecular Meshwork (Review). Int. J. Mol. Med. 2018, 43, 671–681. [Google Scholar] [CrossRef]
- Gupta, V.; Somarajan, B.I.; Gupta, S.; Chaurasia, A.K.; Kumar, S.; Dutta, P.; Gupta, V.; Sharma, A.; Tayo, B.O.; Nischal, K. The Inheritance of Juvenile Onset Primary Open Angle Glaucoma. Clin. Genet. 2017, 92, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Alward, W.L.M.; Fingert, J.H.; Coote, M.A.; Johnson, A.T.; Lerner, S.F.; Junqua, D.; Durcan, F.J.; McCartney, P.J.; Mackey, D.A.; Sheffield, V.C.; et al. Clinical Features Associated with Mutations in the Chromosome 1 Open-Angle Glaucoma Gene (GLC1A). N. Engl. J. Med. 1998, 338, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Lichter, P.R.; Johnson, A.T.; Zhou, Z.; Higashi, M.; Gottfredsdottir, M.; Othman, M.; Moroi, S.E.; Rozsa, F.W.; Schertzer, R.M.; et al. Age-Dependent Prevalence of Mutations at the GLC1A Locus in Primary Open-Angle Glaucoma. Am. J. Ophthalmol. 2000, 130, 165–177. [Google Scholar] [CrossRef]
- Fan, B.J.; Leung, D.Y.L.; Wang, D.Y.; Gobeil, S.; Raymond, V.; Tam, P.O.S.; Lam, D.S.C.; Pang, C.P. Novel Myocilin Mutation in a Chinese Family with Juvenile-Onset Open-Angle Glaucoma. Arch. Ophthalmol. 2006, 124, 102–106. [Google Scholar] [CrossRef]
- Markandaya, M.; Ramesh, T.K.; Selvaraju, V.; Dorairaj, S.K.; Prakash, R.; Shetty, J.; Kumar, A. Genetic Analysis of an Indian Family with Members Affected with Juvenile-Onset Primary Open-Angle Glaucoma. Ophthalmic Genet. 2004, 25, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Svidnicki, P.V.; Braghini, C.A.; Costa, V.P.; Schimiti, R.B.; de Vasconcellos, J.P.C.; de Melo, M.B. Occurrence of MYOC and CYP1B1 Variants in Juvenile Open Angle Glaucoma Brazilian Patients. Ophthalmic Genet. 2018, 39, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Waryah, A.M.; Narsani, A.K.; Sheikh, S.A.; Shaikh, H.; Shahani, M.Y. The Novel Heterozygous Thr377Arg MYOC Mutation Causes Severe Juvenile Open Angle Glaucoma in a Large Pakistani Family. Gene 2013, 528, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Xie, L.; Wu, Z.; Cao, Y.; Zheng, Y.; Pang, C.-P.; Zhang, M. Detection of Mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese Juvenile Onset Open-Angle Glaucoma Using Exome Sequencing. Sci. Rep. 2018, 8, 4498. [Google Scholar] [CrossRef]
- Criscione, J.; Ji, W.; Jeffries, L.; McGrath, J.M.; Soloway, S.; Pusztai, L.; Lakhani, S. Identification of a Novel MYOC Variant in a Hispanic Family with Early-Onset Primary Open-Angle Glaucoma with Elevated Intraocular Pressure. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004374. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Noda, S.; Imamura, Y.; Nagasawa, A.; Kubota, R.; Mashima, Y.; Kudoh, J.; Oguchi, Y.; Shimizu, N. Mouse Myocilin (Myoc) Gene Expression in Ocular Tissues. Biochem. Biophys. Res. Commun. 1998, 248, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Ortego, J.; Escribano, J.; Coca-Prados, M. Cloning and Characterization of Subtracted cDNAs from a Human Ciliary Body Library Encoding TIGR, a Protein Involved in Juvenile Open Angle Glaucoma with Homology to Myosin and Olfactomedin. FEBS Lett. 1997, 413, 349–353. [Google Scholar] [CrossRef]
- Karali, A.; Russell, P.; Stefani, F.H.; Tamm, E.R. Localization of Myocilin/Trabecular Meshwork--Inducible Glucocorticoid Response Protein in the Human Eye. Investig. Ophthalmol. Vis. Sci. 2000, 41, 729–740. [Google Scholar]
- Kubota, R.; Noda, S.; Wang, Y.; Minoshima, S.; Asakawa, S.; Kudoh, J.; Mashima, Y.; Oguchi, Y.; Shimizu, N. A Novel Myosin-like Protein (Myocilin) Expressed in the Connecting Cilium of the Photoreceptor: Molecular Cloning, Tissue Expression, and Chromosomal Mapping. Genomics 1997, 41, 360–369. [Google Scholar] [CrossRef]
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F.; et al. CRISPR-Cas9–Based Treatment of Myocilin-Associated Glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204. [Google Scholar] [CrossRef]
- Wiggs, J.L. Molecular and Clinical Evaluation of a Patient Hemizygous for TIGR/MYOC. Arch. Ophthalmol. 2001, 119, 1674. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Savinova, O.V.; Reedy, M.V.; Martin, J.; Lun, Y.; Gan, L.; Smith, R.S.; Tomarev, S.I.; John, S.W.M.; Johnson, R.L. Targeted Disruption of the Myocilin Gene (Myoc) Suggests That Human Glaucoma-Causing Mutations Are Gain of Function. Mol. Cell. Biol. 2001, 21, 7707–7713. [Google Scholar] [CrossRef]
- Tamm, E.R.; Russell, P.; Epstein, D.L.; Johnson, D.H.; Piatigorsky, J. Modulation of Myocilin/TIGR Expression in Human Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2577–2582. [Google Scholar]
- Carbone, M.A.; Ayroles, J.F.; Yamamoto, A.; Morozova, T.V.; West, S.A.; Magwire, M.M.; Mackay, T.F.C.; Anholt, R.R.H. Overexpression of Myocilin in the Drosophila Eye Activates the Unfolded Protein Response: Implications for Glaucoma. PLoS ONE 2009, 4, e4216. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 301, pp. 215–290. ISBN 978-0-12-407704-1. [Google Scholar]
- Gould, D.B.; Miceli-Libby, L.; Savinova, O.V.; Torrado, M.; Tomarev, S.I.; Smith, R.S.; John, S.W.M. Genetically Increasing Myoc Expression Supports a Necessary Pathologic Role of Abnormal Proteins in Glaucoma. Mol. Cell. Biol. 2004, 24, 9019–9025. [Google Scholar] [CrossRef] [PubMed]
- Shepard, A.R.; Jacobson, N.; Millar, J.C.; Pang, I.-H.; Steely, H.T.; Searby, C.C.; Sheffield, V.C.; Stone, E.M.; Clark, A.F. Glaucoma-Causing Myocilin Mutants Require the Peroxisomal Targeting Signal-1 Receptor (PTS1R) to Elevate Intraocular Pressure. Hum. Mol. Genet. 2007, 16, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Harder, J.M.; Williams, P.A.; Rausch, R.L.; Kiernan, A.E.; Nair, K.S.; Anderson, M.G.; John, S.W.M.; Howell, G.R.; Libby, R.T. Using Genetic Mouse Models to Gain Insight into Glaucoma: Past Results and Future Possibilities. Exp. Eye Res. 2015, 141, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Zhang, D.; Zhang, J.; Sun, H.; Du, Q.; Li, X. Updates on the Molecular Genetics of Primary Congenital Glaucoma (Review). Exp. Ther. Med. 2020, 20, 968–977. [Google Scholar] [CrossRef]
- Gauthier, A.C.; Wiggs, J.L. Childhood Glaucoma Genes and Phenotypes: Focus on FOXC1 Mutations Causing Anterior Segment Dysgenesis and Hearing Loss. Exp. Eye Res. 2020, 190, 107893. [Google Scholar] [CrossRef]
- Fox, A.R.; Fingert, J.H. Familial Normal Tension Glaucoma Genetics. Prog. Retin. Eye Res. 2023, 96, 101191. [Google Scholar] [CrossRef]
- Sowden, J.C. Molecular and Developmental Mechanisms of Anterior Segment Dysgenesis. Eye 2007, 21, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; Walter, M.A. Genomics and Anterior Segment Dysgenesis: A Review. Clin. Exp. Ophthalmol. 2014, 42, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, S.; Acharya, M.; Banerjee, D.; Bhattacharjee, A.; Ray, K. Molecular Basis for Involvement of CYP1B1 in MYOC Upregulation and Its Potential Implication in Glaucoma Pathogenesis. PLoS ONE 2012, 7, e45077. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, C.; Arno, G.; Brookes, J.; Garcia-Feijoo, J.; Khaw, P.T.; Moosajee, M. Primary Congenital Glaucoma Including Next-Generation Sequencing-Based Approaches: Clinical Utility Gene Card. Eur. J. Hum. Genet. 2018, 26, 1713–1718. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Iwata, T. Exploring the Genetic Landscape of Childhood Glaucoma. Children 2024, 11, 454. https://doi.org/10.3390/children11040454
Pan Y, Iwata T. Exploring the Genetic Landscape of Childhood Glaucoma. Children. 2024; 11(4):454. https://doi.org/10.3390/children11040454
Chicago/Turabian StylePan, Yang, and Takeshi Iwata. 2024. "Exploring the Genetic Landscape of Childhood Glaucoma" Children 11, no. 4: 454. https://doi.org/10.3390/children11040454
APA StylePan, Y., & Iwata, T. (2024). Exploring the Genetic Landscape of Childhood Glaucoma. Children, 11(4), 454. https://doi.org/10.3390/children11040454