
Lung Diseases and Rare Disorders: Is It a Lysosomal Storage Disease? Differential Diagnosis, Pathogenetic Mechanisms and Management
 , , , , ,
, , , , ,  , and
, and
Abstract
:
1. Introduction
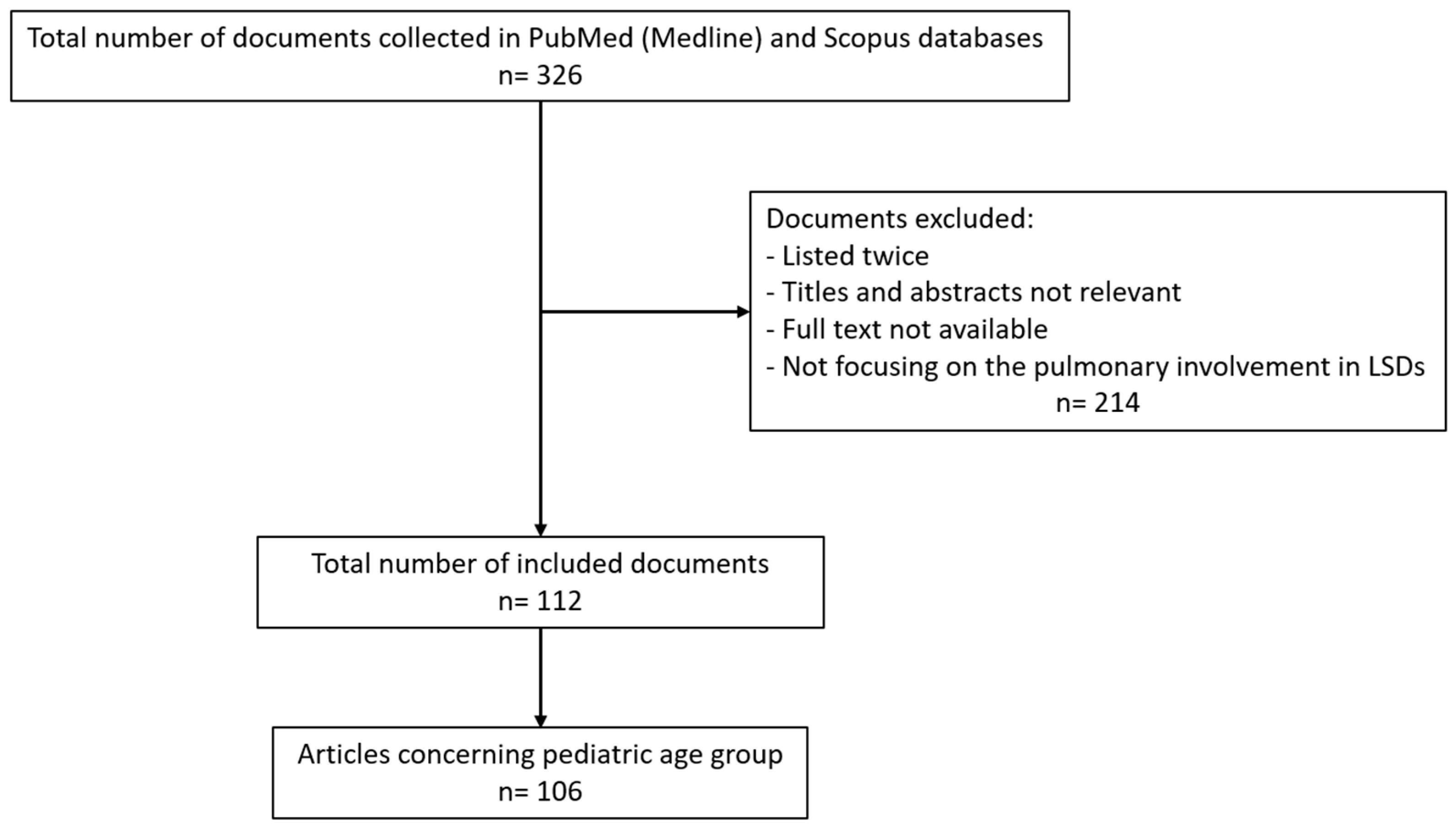
2. Methods
3. Results
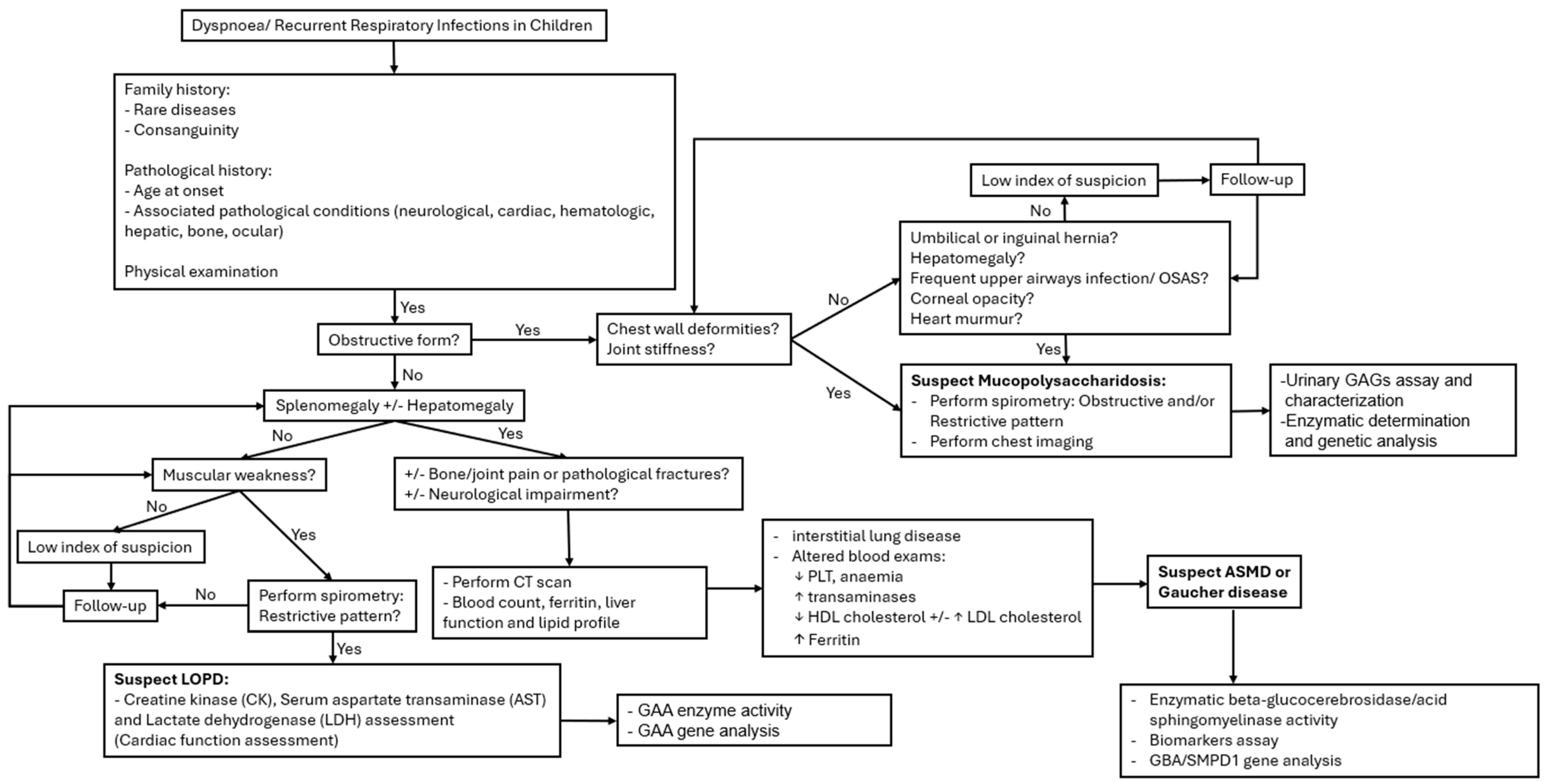
3.1. Clinical Presentation
3.1.1. Primary Respiratory Impairment
3.1.2. Secondary Respiratory Impairment
3.2. Approach to Differential Diagnosis
3.2.1. Radiological and Lung Function Patterns
3.2.2. Clinical Manifestations Associated
3.2.3. Biochemical Abnormalities
3.3. Treatment and Follow-Up
3.3.1. Gaucher Disease
3.3.2. Acid Sphingomyelinase Deficiency
3.3.3. Mucopolysaccharidosis
3.3.4. Pompe Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bajaj, S.; Muranjan, M.; Karande, S.; Prabhat, D. Rare Disease Heralded by Pulmonary Manifestations: Avoiding Pitfalls of an “Asthma” Label. J. Postgrad. Med. 2017, 63, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher Disease Epidemiology and Natural History: A Comprehensive Review of the Literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, R.J.; Velayati, A.; Goldin, E.; Sidransky, E. The Role of Saposin C in Gaucher Disease. Mol. Genet. Metab. 2012, 106, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine Is a Key Biomarker of Gaucher Disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Gawad Tantawy, A.A.; Moneam Adly, A.A.; Madkour, S.S.; Salah El-Din, N.Y. Pulmonary Manifestations in Young Gaucher Disease Patients: Phenotype-genotype Correlation and Radiological Findings. Pediatr. Pulmonol. 2020, 55, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Vellas, D.; Gramont, B.; Grange, R.; Cathébras, P. Pulmonary Involvement Responsive to Enzyme Replacement Therapy in an Elderly Patient with Gaucher Disease. Eur. J. Case Rep. Intern. Med. 2021, 8, 002802. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Caillaud, C.; Levade, T. Disorders of Sphingolipid Synthesis, Sphingolipidoses, Niemann-Pick Disease Type C and Neuronal Ceroid Lipofuscinoses. In Inborn Metabolic Diseases; Saudubray, J.-M., Baumgartner, M.R., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 551–575. ISBN 978-3-662-49769-2. [Google Scholar]
- Pan, Y.-W.; Tsai, M.-C.; Yang, C.-Y.; Yu, W.-H.; Wang, B.; Yang, Y.-J.; Chou, Y.-Y. Enzyme Replacement Therapy for Children with Acid Sphingomyelinase Deficiency in the Real World: A Single Center Experience in Taiwan. Mol. Genet. Metab. Rep. 2023, 34, 100957. [Google Scholar] [CrossRef]
- Cassiman, D.; Packman, S.; Bembi, B.; Turkia, H.B.; Al-Sayed, M.; Schiff, M.; Imrie, J.; Mabe, P.; Takahashi, T.; Mengel, K.E.; et al. Cause of Death in Patients with Chronic Visceral and Chronic Neurovisceral Acid Sphingomyelinase Deficiency (Niemann-Pick Disease Type B and B Variant): Literature Review and Report of New Cases. Mol. Genet. Metab. 2016, 118, 206–213. [Google Scholar] [CrossRef]
- Geberhiwot, T.; Wasserstein, M.; Wanninayake, S.; Bolton, S.C.; Dardis, A.; Lehman, A.; Lidove, O.; Dawson, C.; Giugliani, R.; Imrie, J.; et al. Consensus Clinical Management Guidelines for Acid Sphingomyelinase Deficiency (Niemann–Pick Disease Types A, B and A/B). Orphanet J. Rare Dis. 2023, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Desnick, R.J.; Schuchman, E.H.; Hossain, S.; Wallenstein, S.; Lamm, C.; McGovern, M.M. The Natural History of Type B Niemann-Pick Disease: Results From a 10-Year Longitudinal Study. Pediatrics 2004, 114, e672–e677. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, N.; Troadec, C.; De Villemeur, T.B.; Clément, A.; Fauroux, B. Lung Disease in Niemann–Pick Disease. Pediatr. Pulmonol. 2007, 42, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Capron, T.; Trigui, Y.; Gautier, C.; Puech, B.; Chanez, P.; Reynaud-Gaubert, M. Respiratory Impairment in Niemann-Pick B Disease: Two Case Reports and Review for the Pulmonologist. Respir. Med. Res. 2019, 76, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical Presentation and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The Emerging Phenotype of Late-Onset Pompe Disease: A Systematic Literature Review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- Reuser, A.J.J.; Hirschhorn, R.; Kroos, M.A. Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Santamaria, F.; Montella, S.; Mirra, V.; De Stefano, S.; Andria, G.; Parenti, G. Respiratory Manifestations in Patients with Inherited Metabolic Diseases. Eur. Respir. Rev. 2013, 22, 437–453. [Google Scholar] [CrossRef]
- Faverio, P.; Stainer, A.; De Giacomi, F.; Gasperini, S.; Motta, S.; Canonico, F.; Pieruzzi, F.; Monzani, A.; Pesci, A.; Biondi, A. Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. Int. J. Mol. Sci. 2019, 20, 327. [Google Scholar] [CrossRef]
- Rapoport, D.M.; Mitchell, J.J. Pathophysiology, Evaluation, and Management of Sleep Disorders in the Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122, 49–54. [Google Scholar] [CrossRef]
- Borie, R.; Crestani, B.; Guyard, A.; Lidove, O. Interstitial Lung Disease in Lysosomal Storage Disorders. Eur. Respir. Rev. 2021, 30, 200363. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Sirrs, S.; Chan, A.; Pritzker, M.R.; Duffy, T.P.; Grace, M.E.; Meeker, D.P.; Goldman, M.E. Pulmonary Hypertension in Type 1 Gaucher’s Disease: Genetic and Epigenetic Determinants of Phenotype and Response to Therapy. Mol. Genet. Metab. 2002, 77, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jais, X. Splenectomy and Chronic Thromboembolic Pulmonary Hypertension. Thorax 2005, 60, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.M.; Liu, J.; Chen, F.; Pastores, G.M.; Knowles, J.; Boxer, M.; Aleck, K.; Mistry, P.K. Pulmonary Vascular Disease in Gaucher Disease: Clinical Spectrum, Determinants of Phenotype and Long-term Outcomes of Therapy. J. Inherit. Metab. Dis. 2011, 34, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, F.; Parenti, G.; Guidi, G.; Filocamo, M.; Strisciuglio, P.; Grillo, G.; Farina, V.; Sarnelli, P.; Rizzolo, M.G.; Rotondo, A.; et al. Pulmonary Manifestations of Gaucher Disease: An Increased Risk for L444P Homozygotes? Am. J. Respir. Crit. Care Med. 1998, 157, 985–989. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Wasserstein, M.P.; Giugliani, R.; Bembi, B.; Vanier, M.T.; Mengel, E.; Brodie, S.E.; Mendelson, D.; Skloot, G.; Desnick, R.J.; et al. A Prospective, Cross-Sectional Survey Study of the Natural History of Niemann-Pick Disease Type B. Pediatrics 2008, 122, e341–e349. [Google Scholar] [CrossRef] [PubMed]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s Disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- McGovern, M.M.; Lippa, N.; Bagiella, E.; Schuchman, E.H.; Desnick, R.J.; Wasserstein, M.P. Morbidity and Mortality in Type B Niemann–Pick Disease. Genet. Med. 2013, 15, 618–623. [Google Scholar] [CrossRef]
- Semenza, G.L.; Pyeritz, R.E. Respiratory Complications of Mucopolysaccharide Storage Disorders. Medicine 1988, 67, 209–219. [Google Scholar] [CrossRef]
- Asseri, A.A.; Alzoani, A.; Almazkary, A.M.; Abdulaziz, N.; Almazkary, M.H.; Alahmari, S.A.; Duraisamy, A.J.; Sureshkumar, S. Mucopolysaccharidosis Type I Presenting with Persistent Neonatal Respiratory Distress: A Case Report. Diseases 2023, 11, 67. [Google Scholar] [CrossRef]
- Bush, D.; Sremba, L.; Lomax, K.; Lipsett, J.; Ketteridge, D.; Bratkovic, D.; Enchautegui-Colon, Y.; Weisfeld-Adams, J.; Galambos, C.; Lummus, S.; et al. Neonatal Onset Interstitial Lung Disease as a Primary Presenting Manifestation of Mucopolysaccharidosis Type I. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2018; Volume 43, pp. 71–77. ISBN 978-3-662-58613-6. [Google Scholar]
- Chiang, J.; Raiman, J.; Cutz, E.; Solomon, M.; Dell, S. Tachypnea of Infancy as the First Sign of Sanfilippo Syndrome. Pediatrics 2014, 134, e884–e888. [Google Scholar] [CrossRef] [PubMed]
- Paget, T.L.; Parkinson-Lawrence, E.J.; Trim, P.J.; Autilio, C.; Panchal, M.H.; Koster, G.; Echaide, M.; Snel, M.F.; Postle, A.D.; Morrison, J.L.; et al. Increased Alveolar Heparan Sulphate and Reduced Pulmonary Surfactant Amount and Function in the Mucopolysaccharidosis IIIA Mouse. Cells 2021, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Jezela-Stanek, A.; Chorostowska-Wynimko, J.; Tylki-Szymańska, A. Pulmonary Involvement in Selected Lysosomal Storage Diseases and the Impact of Enzyme Replacement Therapy: A State-of-the Art Review. Clin. Respir. J. 2020, 14, 422–429. [Google Scholar] [CrossRef]
- Harmatz, P. Mucopolysaccharidosis VI Pathophysiology Diagnosis and Treatment. Front. Biosci. 2017, 22, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; De Oliveira Poswar, F.; Michelin-Tirelli, K.; Matte, U.D.S.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Wooten, W.I.; Muenzer, J.; Vaughn, B.V.; Muhlebach, M.S. Relationship of Sleep to Pulmonary Function in Mucopolysaccharidosis II. J. Pediatr. 2013, 162, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Brama, I.; Gay, I.; Feinmesser, R.; Springer, C. Upper Airway Obstruction in Hunter Syndrome. Int. J. Pediatr. Otorhinolaryngol. 1986, 11, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kamin, W. Diagnosis and Management of Respiratory Involvement in Hunter Syndrome. Acta Paediatr. 2008, 97, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Poswar, F.D.O.; Henriques Nehm, J.; Kubaski, F.; Poletto, E.; Giugliani, R. Diagnosis and Emerging Treatment Strategies for Mucopolysaccharidosis VII (Sly Syndrome). Ther. Clin. Risk Manag. 2022, 18, 1143–1155. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the Mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef]
- Buhain, W.J.; Rammohan, G.; Berger, H.W. Pulmonary Function in Morquio’s Disease: A Study of Two Siblings. Chest 1975, 68, 41–45. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010, 5, 5. [Google Scholar] [CrossRef]
- Nagpal, R.; Goyal, R.; Priyadarshini, K.; Kashyap, S.; Sharma, M.; Sinha, R.; Sharma, N. Mucopolysaccharidosis: A Broad Review. Indian J. Ophthalmol. 2022, 70, 2249. [Google Scholar] [CrossRef]
- Wagner, V.F.; Northrup, H. Mucopolysaccharidosis Type III. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- El Haddad, L.; Khan, M.; Soufny, R.; Mummy, D.; Driehuys, B.; Mansour, W.; Kishnani, P.S.; ElMallah, M.K. Monitoring and Management of Respiratory Function in Pompe Disease: Current Perspectives. Ther. Clin. Risk Manag. 2023, 19, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.A.; Padidela, R.; Wilkinson, S. Pulmonary Manifestations of Endocrine and Metabolic Diseases in Children. Pediatr. Clin. N. Am. 2021, 68, 81–102. [Google Scholar] [CrossRef]
- Fuller, D.D.; ElMallah, M.K.; Smith, B.K.; Corti, M.; Lawson, L.A.; Falk, D.J.; Byrne, B.J. The Respiratory Neuromuscular System in Pompe Disease. Respir. Physiol. Neurobiol. 2013, 189, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Niu, D.; Tai, S.; Wang, T.; Su, H.; Huang, L.; Soong, W. Airway Abnormalities in Very Early Treated Infantile-onset Pompe Disease: A Large-scale Survey by Flexible Bronchoscopy. Am. J. Med. Genet. Part A 2020, 182, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Gambetti, P.; Dimauro, S.; Baker, L. Nervous System in Pompe’s Disease: Ultrastructure and Biochemistry. J. Neuropathol. Exp. Neurol. 1971, 30, 412–430. [Google Scholar] [CrossRef]
- Martin, J.J.; De Barsy, T.; Van Hoof, F.; Palladini, G. Pompe’s Disease: An Inborn Lysosomal Disorder with Storage of Glycogen: A Study of Brain and Striated Muscle. Acta Neuropathol. 1973, 23, 229–244. [Google Scholar] [CrossRef]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural Deficits Contribute to Respiratory Insufficiency in Pompe Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe Disease Diagnosis and Management Guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef]
- Gülhan, B.; Özçelik, U.; Gürakan, F.; Güçer, Ş.; Orhan, D.; Cinel, G.; Yalçın, E.; Ersöz, D.D.; Kiper, N.; Yüce, A.; et al. Different Features of Lung Involvement in Niemann-Pick Disease and Gaucher Disease. Respir. Med. 2012, 106, 1278–1285. [Google Scholar] [CrossRef]
- Mendelson, D.S.; Wasserstein, M.P.; Desnick, R.J.; Glass, R.; Simpson, W.; Skloot, G.; Vanier, M.; Bembi, B.; Giugliani, R.; Mengel, E.; et al. Type B Niemann-Pick Disease: Findings at Chest Radiography, Thin-Section CT, and Pulmonary Function Testing. Radiology 2006, 238, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Freitas, H.M.P.; Mançano, A.D.; Rodrigues, R.S.; Hochhegger, B.; Torres, P.P.T.E.S.; Escuissato, D.; Araujo Neto, C.A.; Marchiori, E. Niemann-Pick Disease Type B: HRCT Assessment of Pulmonary Involvement. J. Bras. Pneumol. 2017, 43, 451–455. [Google Scholar] [CrossRef]
- Miller, A.; Brown, L.; Pastores, G.; Desnick, R. Pulmonary Involvement in Type 1 Gaucher Disease: Functional and Exercise Findings in Patients with and without Clinical Interstitial Lung Disease. Clin. Genet. 2003, 63, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Prigent, H.; Orlikowski, D.; Laforêt, P.; Letilly, N.; Falaize, L.; Pellegrini, N.; Annane, D.; Raphael, J.-C.; Lofaso, F. Supine Volume Drop and Diaphragmatic Function in Adults with Pompe Disease. Eur. Respir. J. 2012, 39, 1545–1546. [Google Scholar] [CrossRef]
- Pellegrini, N. Respiratory Insufficiency and Limb Muscle Weakness in Adults with Pompe’s Disease. Eur. Respir. J. 2005, 26, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Spiesshoefer, J.; Henke, C.; Kabitz, H.J.; Brix, T.; Görlich, D.; Herkenrath, S.; Randerath, W.; Young, P.; Boentert, M. The Nature of Respiratory Muscle Weakness in Patients with Late-Onset Pompe Disease. Neuromuscul. Disord. 2019, 29, 618–627. [Google Scholar] [CrossRef]
- Kubaski, F.; Tomatsu, S.; Patel, P.; Shimada, T.; Xie, L.; Yasuda, E.; Mason, R.; Mackenzie, W.G.; Theroux, M.; Bober, M.B.; et al. Non-Invasive Pulmonary Function Test on Morquio Patients. Mol. Genet. Metab. 2015, 115, 186–192. [Google Scholar] [CrossRef]
- Lachman, R.S.; Burton, B.K.; Clarke, L.A.; Hoffinger, S.; Ikegawa, S.; Jin, D.-K.; Kano, H.; Kim, O.-H.; Lampe, C.; Mendelsohn, N.J.; et al. Mucopolysaccharidosis IVA (Morquio A Syndrome) and VI (Maroteaux–Lamy Syndrome): Under-Recognized and Challenging to Diagnose. Skelet. Radiol. 2014, 43, 359–369. [Google Scholar] [CrossRef]
- Shih, S.L.; Lee, Y.J.; Lin, S.P.; Sheu, C.Y.; Blickman, J.G. Airway changes in children with mucopolysaccharidoses. Acta Radiol. 2002, 43, 40–43. [Google Scholar] [CrossRef]
- Lin, S.; Shih, S.; Chuang, C.; Lee, K.; Chen, M.; Niu, D.; Chiu, P.C.; Lin, S.J.; Lin, H. Characterization of Pulmonary Function Impairments in Patients with Mucopolysaccharidoses—Changes with Age and Treatment. Pediatr. Pulmonol. 2014, 49, 277–284. [Google Scholar] [CrossRef]
- Rodriguez, M.E.; Mackenzie, W.G.; Ditro, C.; Miller, T.L.; Chidekel, A.; Shaffer, T.H. Skeletal Dysplasias: Evaluation with Impulse Oscillometry and Thoracoabdominal Motion Analysis. Pediatr. Pulmonol. 2010, 45, 679–686. [Google Scholar] [CrossRef]
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Scarpa, M.; Tobin, M.J.; Ward, S.A.; Rapoport, D.M. Respiratory and Sleep Disorders in Mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef]
- Simmons, M.A.; Bruce, I.A.; Penney, S.; Wraith, E.; Rothera, M.P. Otorhinolaryngological Manifestations of the Mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 589–595. [Google Scholar] [CrossRef]
- Shah, N.M.; Sharma, L.; Ganeshamoorthy, S.; Kaltsakas, G. Respiratory Failure and Sleep-Disordered Breathing in Late-Onset Pompe Disease: A Narrative Review. J. Thorac. Dis. 2020, 12, S235–S247. [Google Scholar] [CrossRef]
- Marques, J.S. The Clinical Management of Pompe Disease: A Pediatric Perspective. Children 2022, 9, 1404. [Google Scholar] [CrossRef]
- McGovern, M.M.; Avetisyan, R.; Sanson, B.-J.; Lidove, O. Disease Manifestations and Burden of Illness in Patients with Acid Sphingomyelinase Deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41. [Google Scholar] [CrossRef]
- Lidove, O.; Sedel, F.; Charlotte, F.; Froissart, R.; Vanier, M.T. Cirrhosis and Liver Failure: Expanding Phenotype of Acid Sphingomyelinase-Deficient Niemann-Pick Disease in Adulthood. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early Diagnostic Signs in Infants and Children. Ital. J. Pediatr. 2018, 44, 133. [Google Scholar] [CrossRef]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early Disease Progression of Hurler Syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef]
- Parini, R.; Jones, S.A.; Harmatz, P.R.; Giugliani, R.; Mendelsohn, N.J. The Natural History of Growth in Patients with Hunter Syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2016, 117, 438–446. [Google Scholar] [CrossRef]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 Gangliosidosis and Morquio B Disease: An Update on Genetic Alterations and Clinical Findings. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Rigoldi, M.; Verrecchia, E.; Manna, R.; Mascia, M.T. Clinical Hints to Diagnosis of Attenuated Forms of Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 132. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Vincent, C.; Fain, O.; Fantin, B.; Mentre, F. Bone Events and Evolution of Biologic Markers in Gaucher Disease before and during Treatment. Arthritis Res. Ther. 2010, 12, R156. [Google Scholar] [CrossRef]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during Type 1 Gaucher Disease: Mechanisms and Progression under Treatment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef]
- Mahalingam, K.; Janani, S.; Priya, S.; Elango, E.M.; Sundari, R.M. Diagnosis of mucopolysaccharidoses: How to avoid false positives and false negatives. Indian J. Pediatr. 2004, 71, 29–32. [Google Scholar] [CrossRef]
- Hobson-Webb, L.D.; DeArmey, S.; Kishnani, P.S. The Clinical and Electrodiagnostic Characteristics of Pompe Disease with Post-Enzyme Replacement Therapy Findings. Clin. Neurophysiol. 2011, 122, 2312–2317. [Google Scholar] [CrossRef]
- Goitein, O. Lung Involvement and Enzyme Replacement Therapy in Gaucher’s Disease. QJM 2001, 94, 407–415. [Google Scholar] [CrossRef]
- Tezuka, Y.; Fukuda, M.; Watanabe, S.; Nakano, T.; Okamoto, K.; Kuzume, K.; Yano, Y.; Eguchi, M.; Ishimae, M.; Ishii, E.; et al. Histological Characterisation of Visceral Changes in a Patient with Type 2 Gaucher Disease Treated with Enzyme Replacement Therapy. Blood Cells Mol. Dis. 2018, 68, 194–199. [Google Scholar] [CrossRef]
- Rao, A.R.; Parakininkas, D.; Hintermeyer, M.; Segura, A.D.; Rice, T.B. Bilateral Lung Transplant in Gauchers Type-1 Disease. Pediatr. Transplant. 2005, 9, 239–243. [Google Scholar] [CrossRef]
- Machaczka, M.; Lorenz, F.; Kleinotiene, G.; Bulanda, A.; Markuszewska-Kuczyńska, A.; Raistenskis, J.; Klimkowska, M. Recurrent Pulmonary Aspergillosis and Mycobacterial Infection in an Unsplenectomized Patient with Type 1 Gaucher Disease. Upsala J. Med. Sci. 2014, 119, 44–49. [Google Scholar] [CrossRef]
- Elstein, D.; Klutstein, M.W.; Lahad, A.; Abrahamov, A.; Hadas-Halpern, I.; Zimran, A. Echocardiographic Assessment of Pulmonary Hypertension in Gaucher’s Disease. Lancet 1998, 351, 1544–1546. [Google Scholar] [CrossRef]
- Jones, S.A.; McGovern, M.; Lidove, O.; Giugliani, R.; Mistry, P.K.; Dionisi-Vici, C.; Munoz-Rojas, M.-V.; Nalysnyk, L.; Schecter, A.D.; Wasserstein, M. Clinical Relevance of Endpoints in Clinical Trials for Acid Sphingomyelinase Deficiency Enzyme Replacement Therapy. Mol. Genet. Metab. 2020, 131, 116–123. [Google Scholar] [CrossRef]
- Wasserstein, M.; Dionisi-Vici, C.; Giugliani, R.; Hwu, W.-L.; Lidove, O.; Lukacs, Z.; Mengel, E.; Mistry, P.K.; Schuchman, E.H.; McGovern, M. Recommendations for Clinical Monitoring of Patients with Acid Sphingomyelinase Deficiency (ASMD). Mol. Genet. Metab. 2019, 126, 98–105. [Google Scholar] [CrossRef]
- Hollak, C.E.M.; De Sonnaville, E.S.V.; Cassiman, D.; Linthorst, G.E.; Groener, J.E.; Morava, E.; Wevers, R.A.; Mannens, M.; Aerts, J.M.F.G.; Meersseman, W.; et al. Acid Sphingomyelinase (Asm) Deficiency Patients in The Netherlands and Belgium: Disease Spectrum and Natural Course in Attenuated Patients. Mol. Genet. Metab. 2012, 107, 526–533. [Google Scholar] [CrossRef]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-Year Results of a Clinical Trial of Olipudase Alfa Enzyme Replacement Therapy in Pediatric Patients with Acid Sphingomyelinase Deficiency. Genet. Med. 2021, 23, 1543–1550. [Google Scholar] [CrossRef]
- Sanofi. Positive Topline Results Demonstrated by Olipudase Alfa, First and Only Investigational Therapy in Late-Stage Development for Acid Sphingomyelinase Deficiency. Press Release. Available online: https://www.sanofi.com/en/media-room/press-releases/2020/2020-01-30-06-00-00-1977201 (accessed on 30 January 2024).
- Mannem, H.; Kilbourne, S.; Weder, M. Lung Transplantation in a Patient with Niemann–Pick Disease. J. Heart Lung Transplant. 2019, 38, 100–101. [Google Scholar] [CrossRef]
- O’Neill, R.S.; Belousova, N.; Malouf, M.A. Pulmonary Type B Niemann-Pick Disease Successfully Treated with Lung Transplantation. Case Rep. Transplant. 2019, 2019, 9431751. [Google Scholar] [CrossRef]
- Ding, F.; Mehta, A.C.; Arrossi, A.V. Successful Lung Transplantation in a Patient with Niemann–Pick Disease. J. Heart Lung Transplant. 2019, 38, 582–583. [Google Scholar] [CrossRef]
- Lidove, O.; Le Fèvre, L.; Goasguen, N.; Jamali, M.; Vercellino, L.; Garnier, M.; Khellaf, M.; Belmatoug, N.; Ziza, J.-M. Déficit en sphingomyélinase acide et rupture splénique: Splénectomie ou traitement conservateur ? La Rev. De Med. Interne 2015, 36, 619–622. [Google Scholar] [CrossRef]
- Suarez-Guerrero, J.L.; Gómez Higuera, P.J.I.; Arias Flórez, J.S.; Contreras-García, G.A. Mucopolisacaridosis: Características clínicas, diagnóstico y de manejo. Rev. Chil. De Pediatría 2016, 87, 295–304. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Wijburg, F.A. Therapy for the Mucopolysaccharidoses. Rheumatology 2011, 50, v49–v59. [Google Scholar] [CrossRef]
- McBride, K.L.; Flanigan, K.M. Update in the Mucopolysaccharidoses. Semin. Pediatr. Neurol. 2021, 37, 100874. [Google Scholar] [CrossRef]
- Tulebayeva, A.; Sharipova, M.; Boranbayeva, R. Respiratory Dysfunction in Children and Adolescents with Mucopolysaccharidosis Types I, II, IVA, and VI. Diagnostics 2020, 10, 63. [Google Scholar] [CrossRef]
- Rutten, M.; Ciet, P.; Van Den Biggelaar, R.; Oussoren, E.; Langendonk, J.G.; Van Der Ploeg, A.T.; Langeveld, M. Severe Tracheal and Bronchial Collapse in Adults with Type II Mucopolysaccharidosis. Orphanet J. Rare Dis. 2016, 11, 50. [Google Scholar] [CrossRef]
- Kenth, J.J.; Thompson, G.; Fullwood, C.; Wilkinson, S.; Jones, S.; Bruce, I.A. The Characterisation of Pulmonary Function in Patients with Mucopolysaccharidoses IVA: A Longitudinal Analysis. Mol. Genet. Metab. Rep. 2019, 20, 100487. [Google Scholar] [CrossRef]
- American Association of Neuromuscular & Electrodiagnostic Medicine. Diagnostic Criteria for Late-Onset (Childhood and Adult) Pompe Disease. Muscle Nerve 2009, 40, 149–160. [Google Scholar] [CrossRef]
- Ward, N.S.; Hill, N.S. Pulmonary Function Testing in Neuromuscular Disease. Clin. Chest. Med. 2001, 22, 769–781. [Google Scholar] [CrossRef]
- American Thoracic Society/European Respiratory Society. ATS/ERS Statement on Respiratory Muscle Testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [Google Scholar] [CrossRef]
- van der Ploeg, A.T. Monitoring of Pulmonary Function in Pompe Disease: A Muscle Disease with New Therapeutic Perspectives. Eur. Respir. J. 2005, 26, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Wokke, J.H.J.; Escolar, D.M.; Pestronk, A.; Jaffe, K.M.; Carter, G.T.; van den Berg, L.H.; Florence, J.M.; Mayhew, J.; Skrinar, A.; Corzo, D.; et al. Clinical Features of Late-Onset Pompe Disease: A Prospective Cohort Study. Muscle Nerve 2008, 38, 1236–1245. [Google Scholar] [CrossRef]
- Cupler, E.J.; Berger, K.I.; Leshner, R.T.; Wolfe, G.I.; Han, J.J.; Barohn, R.J.; Kissel, J.T.; Aanem Consensus Committee on Late-Onset Pompe Disease. Consensus Treatment Recommendations for Late-onset Pompe Disease. Muscle Nerve 2012, 45, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Barbey, F.; Lazor, R.; Bonafé, L. Pulmonary Involvement in Adult Patients with Inborn Errors of Metabolism. Respiration 2017, 94, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Praetorius, S.; Karabul, N.; Dießel, J.; Schmidt, D.; Motz, R.; Haase, C.; Baethmann, M.; Hennermann, J.B.; Smitka, M.; et al. Outcome of Patients with Classical Infantile Pompe Disease Receiving Enzyme Replacement Therapy in Germany. In JIMD Reports; Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; Volume 20, pp. 65–75. ISBN 978-3-662-46699-5. [Google Scholar]
- Pena, L.D.M.; Barohn, R.J.; Byrne, B.J.; Desnuelle, C.; Goker-Alpan, O.; Ladha, S.; Laforêt, P.; Mengel, K.E.; Pestronk, A.; Pouget, J.; et al. Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Exploratory Efficacy of the Novel Enzyme Replacement Therapy Avalglucosidase Alfa (neoGAA) in Treatment-Naïve and Alglucosidase Alfa-Treated Patients with Late-Onset Pompe Disease: A Phase 1, Open-Label, Multicenter, Multinational, Ascending Dose Study. Neuromuscul. Disord. 2019, 29, 167–186. [Google Scholar] [CrossRef]
- Dimachkie, M.M.; Barohn, R.J.; Byrne, B.; Goker-Alpan, O.; Kishnani, P.S.; Ladha, S.; Laforêt, P.; Mengel, K.E.; Peña, L.D.M.; Sacconi, S.; et al. Long-Term Safety and Efficacy of Avalglucosidase Alfa in Patients with Late-Onset Pompe Disease. Neurology 2022, 99, E536–E548. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| MPS | Name | Inheritance Pattern | Enzyme Deficiency | Gene |
|---|---|---|---|---|
| I | Hurler, Hurler-Scheie, Scheie | Autosomal Recessive | α-L-iduronidase | IDUA |
| II | Hunter | X-linked Recessive | Iduronate-2-sulfatase | IDS |
| IIIA | Sanfilippo A | Autosomal Recessive | Heparan N-sulphatase | SGHS |
| IIIB | Sanfilippo B | Autosomal Recessive | N-acetyl-α-glucosaminidase | NAGLU |
| IIIC | Sanfilippo C | Autosomal Recessive | α-Glucosaminidase acetyltransferase | HGSNAT |
| IIID | Sanfilippo D | Autosomal Recessive | N-acetylglucosamine-6-sulfatase | GNS |
| IVA | Morquio A | Autosomal Recessive | Galactose-6-sulfate sulfatase | GALNS |
| IVB | Morquio B | Autosomal Recessive | β-galactosidase | GLB1 |
| VI | Maroteauz-Lamy | Autosomal Recessive | N-acetylgalactosamine-4-sulfatase | ARSB |
| VII | Sly | Autosomal Recessive | β-D-glucuronidase | GUS |
| IX | Natowicz | Autosomal Recessive | Hyaluronoglucosaminidase 1 | HYAL1 |
| X | Arylsulfatase K deficiency | Autosomal Recessive | Arylsulfatase K | ARSK |
| “Gaucher Disease” | AND | “Lung disease” OR “Pulmonary involvement” OR “Respiratory symptoms” AND “Diagnosis” OR “Management” OR “Treatment” OR “Follow up” |
| “Acid Sphingomyelinase deficiency” | ||
| “Pompe disease” | ||
| “Mucopolysaccharidosis” |
| Disease | System/Organ Involved | Clinical Manifestations |
|---|---|---|
| Gaucher disease | Liver and spleen | Hepato-splenomegaly |
| Bone marrow | Anemia and thrombocytopenia with easy bruising and bleeding, bone pain | |
| Kidney | Glomerulopathy | |
| Musculoskeletal system | Osteopenia, growth restriction, fractures | |
| Central nervous system | Severe neurodegeneration typically in type 2, slow horizontal saccades typically in type 3 | |
| Acid sphingomyelinase deficiency | Liver and spleen | Hepato-splenomegaly, liver dysfunction |
| Musculoskeletal system | Joint/limb pain and growth delay | |
| Eyes | Cherry-red maculae | |
| Heart | Left ventricular hypertrophy, conduction abnormalities and valvulopathies | |
| Central nervous system (infantile neurovisceral and chronic neurovisceral ASMD) | From mild hypotonia or hyporeflexia to loss of motor ability and mental impairment | |
| Mucopolysaccharidoses | Musculoskeletal system | Skeletal deformities, joint rigidity, growth retardation, coarse faces, gibbus, laxity and flexibility of joints, even though hips and shoulder may be stiffer, hernias |
| Central nervous system | Abnormal behavioral patterns, hypotonia, cognitive impairment, progressive cognitive decline, epilepsy | |
| Liver | Hepatomegaly | |
| Heart | Heart murmur, valvopathy | |
| Eye | Corneal clouding | |
| Ear | Hearing loss | |
| Pompe disease | Musculoskeletal system | Hypotonia, myalgia, asthenia, exercise intolerance and elevated creatine kinase, osteoporosis, scoliosis, rigid spine syndrome |
| Nervous System | Small-fiber neuropathy, sensorineural hearing loss | |
| Oral district | Macroglossia, lingual weakness with dysarthria and dysphagia, failure to thrive | |
| Heart | Cardiac hypertrophy or abnormal cardiac rhythm | |
| Pelvic district | Lower urinary tract (LUT) and anal sphincter involvement | |
| Gastrointestinal (GI) system | Impaired gastric function and GI motility |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montanari, C.; Tagi, V.M.; D’Auria, E.; Guaia, V.; Di Gallo, A.; Ghezzi, M.; Verduci, E.; Fiori, L.; Zuccotti, G. Lung Diseases and Rare Disorders: Is It a Lysosomal Storage Disease? Differential Diagnosis, Pathogenetic Mechanisms and Management. Children 2024, 11, 668. https://doi.org/10.3390/children11060668
Montanari C, Tagi VM, D’Auria E, Guaia V, Di Gallo A, Ghezzi M, Verduci E, Fiori L, Zuccotti G. Lung Diseases and Rare Disorders: Is It a Lysosomal Storage Disease? Differential Diagnosis, Pathogenetic Mechanisms and Management. Children. 2024; 11(6):668. https://doi.org/10.3390/children11060668
Chicago/Turabian StyleMontanari, Chiara, Veronica Maria Tagi, Enza D’Auria, Vincenzo Guaia, Anna Di Gallo, Michele Ghezzi, Elvira Verduci, Laura Fiori, and Gianvincenzo Zuccotti. 2024. "Lung Diseases and Rare Disorders: Is It a Lysosomal Storage Disease? Differential Diagnosis, Pathogenetic Mechanisms and Management" Children 11, no. 6: 668. https://doi.org/10.3390/children11060668