
Characterization of Primary IGF-1 Deficiency in a Cohort of Canadian Children with Short Stature Using a Novel Algorithm Tailored to Electronic Medical Records

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Population
2.3. Identification of Children with SPIGFD
2.4. Statistical Analysis
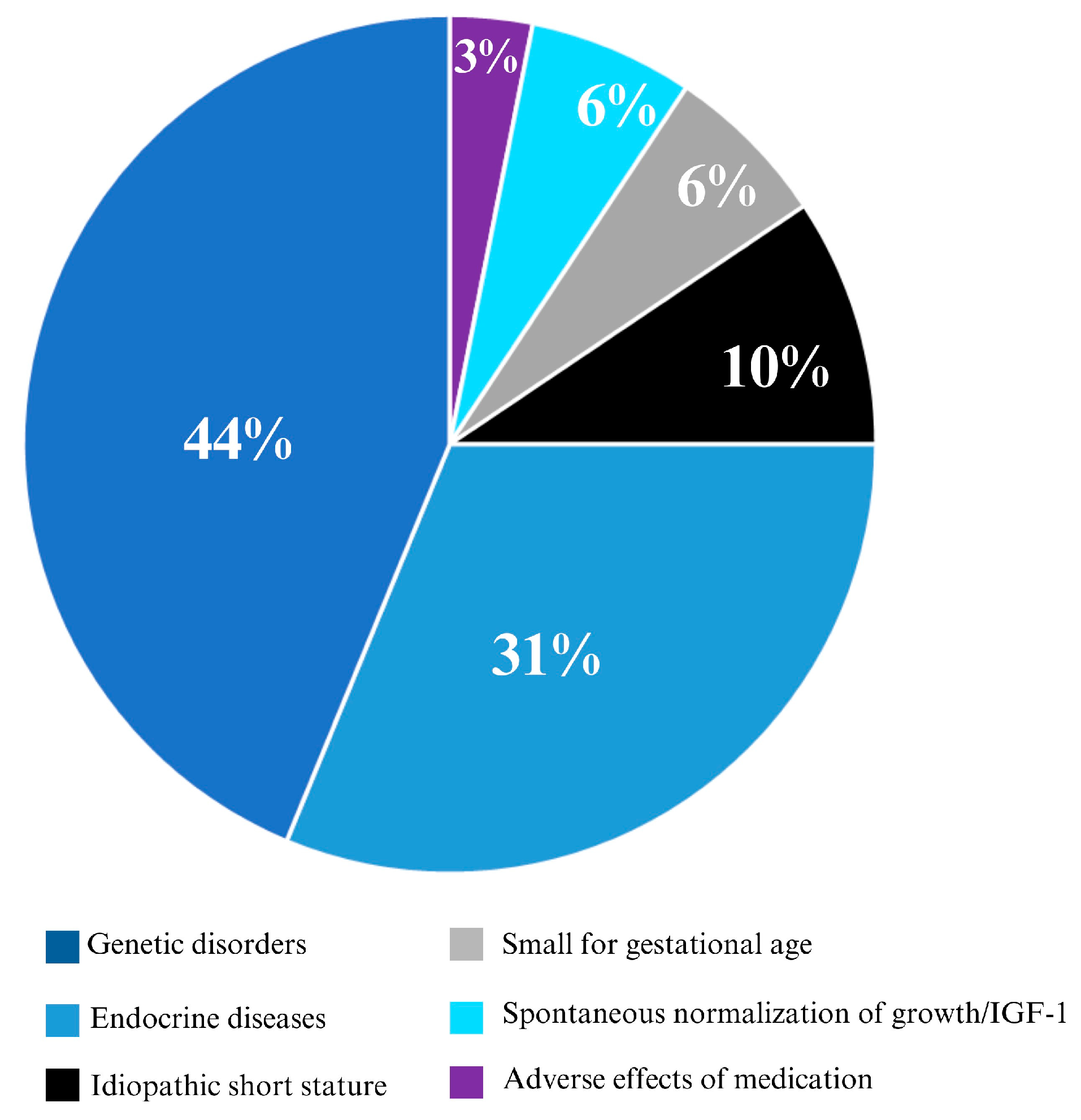
3. Results
3.1. Study Cohort
3.2. Description of Patients with Potential SPIGFD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Study ID | Diagnosis | Sex (M/F) | Age (years) | Height (SD) | Weight (SD) | GH Stimulation Test a | Follow-Up Duration (years) | Change in Height (SDS) |
|---|---|---|---|---|---|---|---|---|
| IGF01 | idiopathic short stature | F | 15.5 | −4.8 | −3.3 | Not performed | 2.6 | 0.7 |
| IGF02 | Turner syndrome (non mosaic Turner syndrome variant) | F | 15.0 | −3.5 | −1.7 | Not performed | 5.6 | 0.2 |
| b IGF04 ♦ | constitutional delay | F | 12.4 | −3.1 | −2.7 | Normal | 7.5 | 1.4 |
| IGF05 ♦ | idiopathic short stature + component of constitutional delay | M | 13.7 | −3.0 | −1.0 | Normal | 4.1 | 0.4 |
| c IGF08 ♢ | familial short stature + component of constitutional delay | M | 13.2 | −4.0 | −2.5 | Normal | 4.1 | 1.1 |
| IGF14 | DMD on chronic glucocorticoids | M | 8.5 | −3.2 | −0.7 | Not performed | 6.2 | −1.3 |
| IGF15 ♦ | genetic condition: 22q11 deletion syndrome | M | 8.3 | −3.0 | −2.4 | Normal | 6.3 | 1.1 |
| IGF17 | cleidocranial dysostosis | M | 6.0 | −3.4 | −1.3 | Not performed | 7.1 | 1.4 |
| IGF19 ♦ | idiopathic short stature | F | 5.8 | −3.5 | −3.0 | Not performed | 6.9 | 0.6 |
| IGF20 ♦ | idiopathic short stature + component of constitutional delay + component of familial short stature | F | 5.4 | −3.6 | −2.4 | Normal | 7.2 | 0.9 |
| IGF22 ♦ | idiopathic short stature + component of constitutional delay + component of familial short stature, responded to GH | F | 5.6 | −3.1 | −1.7 | Normal | 6.5 | 1.1 |
| IGF24 ♦ | idiopathic short stature born SGA | M | 6.7 | −3.2 | −2.7 | Normal | 4.5 | 2.5 |
| IGF26 | septic optic dysplasia | F | 4.8 | −3.6 | −2.1 | Not performed | 6.1 | 0.9 |
| IGF27 ♦ | genetic condition: caspase 8 deficiency | F | 12.4 | −3.1 | −1.9 | Normal | 6.1 | 3.0 |
| IGF28 | genetic condition: phelan-mcdermid syndrome | F | 2.4 | −3.5 | +0.2 | Not performed | 4.8 | −0.5 |
| IGF32 | height spontaneously corrected | M | 2.0 | −3.4 | −0.8 | Not performed | 4.4 | 2.7 |
| IGF33 | familial short stature & growth improved after QVAR d/c | M | 3.1 | −3.4 | −2.2 | Not performed | 6.4 | 1.8 |
| IGF35 | McCune albright, no short stature currently–prev. did have a measurement −3std | M | 3.7 | −3.7 | −4.0 | Not performed | 5.3 | 3.1 |
| IGF36 | septic optic dysplasia | F | 3.9 | −3.1 | −2.7 | Not performed | 4.8 | 0.3 |
| IGF37 ♦ | constitutional delay-hx consistent, spontaneous improvement in growth velocity | M | 12.0 | −3.0 | −1.7 | Normal | 4.4 | 0.8 |
| IGF39 | genetic: hemihypertrophy, normal growth velocity caught up to 3rd percentile; one measurement error prior to age 2 −3 std dev | F | 2.2 | −3.6 | −1.0 | Not performed | 6.3 | 1.8 |
| IGF40 | genetic: 40 megabase deletion chromosome 13 | F | 0.7 | −4.1 | −3.1 | Not performed | 7.4 | 1.4 |
| IGF41 | Turner Syndrome | F | 3.5 | −3.1 | −3.0 | Not performed | 7.1 | −0.6 |
| IGF43 ♦ | idiopathic short stature/IUGR | M | 6.1 | −4.1 | −2.3 | Normal | 0.5 | 1.2 |
| IGF44 | chronic glucocorticoids for autoimmune hemolytic anemia, disproportionate short stature | M | 2.0 | −3.4 | −1.1 | Not performed | 5.6 | −0.4 |
| IGF45 ♦ | genetic: SHOX deletion | M | 16.4 | −4.7 | −3.3 | Normal | 3.7 | −0.3 |
| IGF46 ♦ | spina bifida | M | 3.0 | −3.9 | +3.6 | Normal | 5.7 | −8.2 |
| IGF49 ♦ | constitutional delay | M | 13.5 | −3.2 | −2.6 | Normal | 2.3 | −0.1 |
| IGF50 ♦ | idiopathic short stature | F | 0.7 | −3.4 | −3.7 | Normal | 5.4 | −0.4 |
| IGF51 | nephrogenic DI-x linked, height improved spontaneously, 2017 ht and IGF-1 level meeting criteria | M | 1.4 | −3.8 | −2.4 | Not performed | 4.0 | 2.5 |
Appendix B
- Complete list of the ICD-10 codes used to define secondary causes of IGF-1 deficiency:
- Malnutrition-related ICD-10 codes
- E40→Kwashiorkor
- E41→Nutritional marasmus
- E42→Marasmic kwashiorkor
- E43→Unspecified severe protein-energy malnutrition
- E44→Protein-energy malnutrition of moderate and mild degree
- E44.0→Moderate protein-energy malnutrition
- E44.1→Mild protein-energy malnutrition
- E45→Retarded development following protein-energy malnutrition
- E46→Unspecified protein-energy malnutrition
- Hypothyroidism-related ICD-10 codes
- E03.0→Congenital hypothyroidism with diffuse goitre
- E03.1→Congenital hypothyroidism without goitre
- E03.2→Hypothyroidism due to medicaments and other exogenous substances
- E03.3→Postinfectious hypothyroidism
- E03.5→Myxoedema coma
- E03.8→Other specified hypothyroidism
- E03.9→Hypothyroidism, unspecified
- Growth Hormone Deficiency-related ICD-10 codes
- E23.0→Hypopituitarism
- Disease of Liver-related ICD-10 codes
- Q44.6→Cystic disease of liver
- K70.0→Alcoholic fatty liver
- K70.1→Alcoholic hepatitis
- K70.2→Alcoholic fibrosis and sclerosis of liver
- K70.3→Alcoholic cirrhosis of liver
- K70.4→Alcoholic hepatic failure
- K70.9→Alcoholic liver disease, unspecified
- K71.0→Toxic liver disease with cholestasis
- K71.1→Toxic liver disease with hepatic necrosis
- K71.2→Toxic liver disease with acute hepatitis
- K71.3→Toxic liver disease with chronic persistent hepatitis
- K71.4→Toxic liver disease with chronic lobular hepatitis
- K71.5→Toxic liver disease with chronic active hepatitis
- K71.6→Toxic liver disease with hepatitis, not elsewhere classified
- K71.7→Toxic liver disease with fibrosis and cirrhosis of liver
- K71.8→Toxic liver disease with other disorders of liver
- K71.9→Toxic liver disease, unspecified
- K72.0→Acute and subacute hepatic failure
- K72.1→Chronic hepatic failure
- K72.9→Hepatic failure, unspecified
- K73.0→Chronic persistent hepatitis, not elsewhere classified
- K73.1→Chronic lobular hepatitis, not elsewhere classified
- K73.2→Chronic active hepatitis, not elsewhere classified
- K73.8→Other chronic hepatitis, not elsewhere classified
- K73.9→Chronic hepatitis, unspecified
- K74.0→Hepatic fibrosis
- K74.1→Hepatic sclerosis
- K74.2→Hepatic fibrosis with hepatic sclerosis
- K74.3→Primary biliary cirrhosis
- K74.4→Secondary biliary cirrhosis
- K74.5→Biliary cirrhosis, unspecified
- K74.6→Other and unspecified cirrhosis of liver
- K75.0→Abscess of liver
- K75.2→Nonspecific reactive hepatitis
- K75.3→Granulomatous hepatitis, not elsewhere classified
- K75.4→Autoimmune hepatitis
- K75.8→Other specified inflammatory liver diseases
- K75.9→Inflammatory liver disease, unspecified
- K76.0→Fatty (change of) liver, not elsewhere classified
- K76.1→Chronic passive congestion of liver
- K76.2→Central haemorrhagic necrosis of liver
- K76.3→Infarction of liver
- K76.4→Peliosis hepatis
- K76.7→Hepatorenal syndrome
- K76.8→Other specified diseases of liver
- K76.9→Liver disease, unspecified
- K77.0→Liver disorders in infectious and parasitic diseases classified elsewhere
- K77.8→Liver disorders in other diseases classified elsewhere
References
- Backeljauw, P.F.; Kuntze, J.; Frane, J.; Calikoglu, A.S.; Chernausek, S.D. Adult and Near-Adult Height in Patients with Severe Insulin-Like Growth Factor-I Deficiency after Long-Term Therapy with Recombinant Human Insulin-Like Growth Factor-I. Horm. Res. Paediatr. 2013, 80, 47–56. [Google Scholar] [CrossRef]
- Backeljauw, P.F.; Chernausek, S.D. Treatment of Insulin-Like Growth Factor Deficiency with IGF-I: Studies in Humans. Horm. Res. 2006, 65, 21–27. [Google Scholar] [CrossRef]
- Moore, B.; Whitehead, A.; Davies, K. Short stature, growth hormone deficiency, and primary IGF-1 deficiency. In Advanced Practice in Endocrinology Nursing; Llahana, S., Follin, C., Yedinak, C., Grossman, A., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 13–37. [Google Scholar] [CrossRef]
- Laron, Z. Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef]
- Chernausek, S.D.; Backeljauw, P.F.; Frane, J.; Kuntze, J.; Underwood, L.E. Long-Term Treatment with Recombinant Insulin-Like Growth Factor (IGF)-I in Children with Severe IGF-I Deficiency due to Growth Hormone Insensitivity. J. Clin. Endocrinol. Metab. 2007, 92, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Blethen, S.; Kuntze, J.; Smith, S.L.; Lomax, K.G.; Mathew, P.M. Managing the Child with Severe Primary Insulin-Like Growth Factor-1 Deficiency (IGFD): IGFD Diagnosis and Management. Drugs R D 2014, 14, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Bang, P.; Woelfle, J.; Perrot, V.; Sert, C.; Polak, M. Effectiveness and Safety of rhIGF1 Therapy in Patients with or without Laron Syndrome. Eur. J. Endocrinol. 2021, 184, 267–276. [Google Scholar] [CrossRef]
- Ranke, M.B. Defining Insulin-Like Growth Factor-I Deficiency. Horm. Res. 2006, 65, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Teissier, R.; Flechtner, I.; Colmenares, A.; Lambot-Juhan, K.; Baujat, G.; Pauwels, C.; Samara-Boustani, D.; Beltrand, J.; Simon, A.; Thalassinos, C.; et al. Characterization and Prevalence of Severe Primary IGF1 Deficiency in a Large Cohort of French Children with Short Stature. Eur. J. Endocrinol. 2014, 170, 847–854. [Google Scholar] [CrossRef]
- Edouard, T.; Grünenwald, S.; Gennero, I.; Salles, J.P.; Tauber, M. Prevalence of IGF1 Deficiency in Prepubertal Children with Isolated Short Stature. Eur. J. Endocrinol. 2009, 161, 43–50. [Google Scholar] [CrossRef]
- Kemp, S.F. Insulin-Like Growth Factor-I Deficiency in Children with Growth Hormone Insensitivity: Current and Future Treatment Options. BioDrugs 2009, 23, 155–163. [Google Scholar] [CrossRef]
- WHO. WHO Growth Charts for Canada 2014. Available online: https://www.dietitians.ca/Secondary-Pages/Public/Who-Growth-Charts.aspx (accessed on 3 March 2023).
- Collett-Solberg, P.F.; Misra, M.; Society TCotLWPE. The Role of Recombinant Human Insulin-Like Growth Factor-I in Treating Children with Short Stature. J. Clin. Endocrinol. Metab. 2008, 93, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.M.; Whitehouse, R.H. Clinical Longitudinal Standards for Height, Weight, Height Velocity, Weight Velocity, and Stages of Puberty. Arch. Dis. Child. 1976, 51, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Greulich, W.W.; Pyle, S.I. Radiographic Atlas of Skeletal Development of the Hand and Wrist; Stanford University Press: Redwood City, CA, USA, 1959. [Google Scholar]
- Brämer, G.R. International Statistical Classification of Diseases and Related Health Problems. Tenth Revision. World Health Stat. Q. 1988, 41, 32–36. [Google Scholar] [PubMed]
- Ranke, M.B.; Lindberg, A.; Cowell, C.T.; Wikland, K.A.; Reiter, E.O.; Wilton, P.; Price, D.A.; KIGS International Board. Prediction of Response to Growth Hormone Treatment in Short Children Born Small for Gestational Age: Analysis of Data from KIGS (Pharmacia International Growth Database). J. Clin. Endocrinol. Metab. 2003, 88, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Attie, K.M.; Julius, J.R.; Stoppani, C.; Rundle, A.C. National Cooperative Growth Study Substudy VI: The Clinical Utility of Growth-Hormone-Binding Protein, Insulin-Like Growth Factor I, and Insulin-Like Growth Factor-Binding Protein 3 Measurements. J. Pediatr. 1997, 131 Pt 2, S56–S60. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Ayoola, O.; Whatmore, A.J. Patient Selection for IGF-I Therapy. Horm. Res. 2006, 65 (Suppl. 1), 28–34. [Google Scholar] [CrossRef] [PubMed]
- Frystyk, J.; Freda, P.; Clemmons, D.R. The Current Status of IGF-I Assays—A 2009 Update. Growth Horm. IGF Res. 2010, 20, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, A.; Camacho-Hübner, C.; Woods, K.; Martinelli, C.; Duquesnoy, P.; Savage, M. The Insulin-Like Growth Factor I Generation Test in the Investigation of Short Stature. Acta Paediatr. 1994, 83, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A.; DiVall, S.A.; Polychronakos, C.; Allen, D.B.; Cohen, L.E.; Quintos, J.B.; Rossi, W.C.; Feudtner, C.; Murad, M.H.; Drug and Therapeutics Committee and Ethics Committee of the Pediatric Endocrine Society. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm. Res. Paediatr. 2016, 86, 361–397. [Google Scholar] [CrossRef]
- Zafari, H.; Kosowan, L.; Zulkernine, F.; Signer, A. Diagnosing Post-Traumatic Stress Disorder Using Electronic Medical Record Data. Health Inf. J. 2021, 27, 14604582211053259. [Google Scholar] [CrossRef]
- Slaby, I.; Hain, H.S.; Abrams, D.; Mentch, F.D.; Glessner, J.T.; Sleiman, P.M.A.; Hakonarson, H. An Electronic Health Record (EHR) Phenotype Algorithm to Identify Patients with Attention Deficit Hyperactivity Disorders (ADHD) and Psychiatric Comorbidities. J. Neurodev. Disord. 2022, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.; Johnson, M.E.; Denning, D.W.; Ferreira, G.L.; Cassidy, A. Identifying Rare Diseases Using Electronic Medical Records: The Example of Allergic Bronchopulmonary Aspergillosis. Pharmacoepidemiol. Drug Saf. 2017, 26, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Garcelon, N.; Burgun, A.; Salomon, R.; Neuraz, A. Electronic Health Records for the Diagnosis of Rare Diseases. Kidney Int. 2020, 97, 676–686. [Google Scholar] [CrossRef] [PubMed]

| Study ID | Clinical Diagnosis | GH Treatment (Y/N) | GH Laboratory * and Clinical Treatment Response | Spontaneous Normalization of Growth (Y/N) | Spontaneous Normalization of IGF-1 (Y/N) |
|---|---|---|---|---|---|
| IGF22 | ISS, constitutional delay, familial short stature | Y | Good laboratory and clinical response | N | N |
| IGF40 | Genetic condition: 40 megabase deletion on chromosome 13 | N | N/A | N | Y |
| IGF43 | ISS, intrauterine growth restriction | Y | Good laboratory and clinical response | N | N |
| IGF50 | ISS | N | N/A | N | Y |
| IGF05 | ISS, constitutional delay | N | N/A | N | Y |
| IGF08 | Familial short stature, constitutional delay | Y | Good laboratory response; poor clinical response | N | N |
| IGF49 | Constitutional delay | N | N/A | N | Y |
| IGF37 | Constitutional delay, transient prepubertal slowing of growth with resumption of normal growth velocity at puberty onset | N | N/A | Y | Y |
| IGF19 | ISS | Y | Good laboratory and clinical response | N | N |
| IGF20 | ISS, constitutional delay, familial short stature | Y | Good laboratory and clinical response | N | N |
| IGF39 | Hemihypertrophy syndrome | N | N/A | Y | Y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haridas, R.; Baxter, C.; Dover, S.; Goldbloom, E.B.; Terekhov, I.; Robinson, M.-E. Characterization of Primary IGF-1 Deficiency in a Cohort of Canadian Children with Short Stature Using a Novel Algorithm Tailored to Electronic Medical Records. Children 2024, 11, 727. https://doi.org/10.3390/children11060727
Haridas R, Baxter C, Dover S, Goldbloom EB, Terekhov I, Robinson M-E. Characterization of Primary IGF-1 Deficiency in a Cohort of Canadian Children with Short Stature Using a Novel Algorithm Tailored to Electronic Medical Records. Children. 2024; 11(6):727. https://doi.org/10.3390/children11060727
Chicago/Turabian StyleHaridas, Rinila, Carly Baxter, Saunya Dover, Ellen B. Goldbloom, Ivan Terekhov, and Marie-Eve Robinson. 2024. "Characterization of Primary IGF-1 Deficiency in a Cohort of Canadian Children with Short Stature Using a Novel Algorithm Tailored to Electronic Medical Records" Children 11, no. 6: 727. https://doi.org/10.3390/children11060727
APA StyleHaridas, R., Baxter, C., Dover, S., Goldbloom, E. B., Terekhov, I., & Robinson, M.-E. (2024). Characterization of Primary IGF-1 Deficiency in a Cohort of Canadian Children with Short Stature Using a Novel Algorithm Tailored to Electronic Medical Records. Children, 11(6), 727. https://doi.org/10.3390/children11060727