
FLAIR Hyperintense Cortical Lesions in a 4-Year-Old Child with Myelin Oligodendrocyte Glycoprotein (MOG)-Associated Encephalitis and Seizures: A Case Report
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
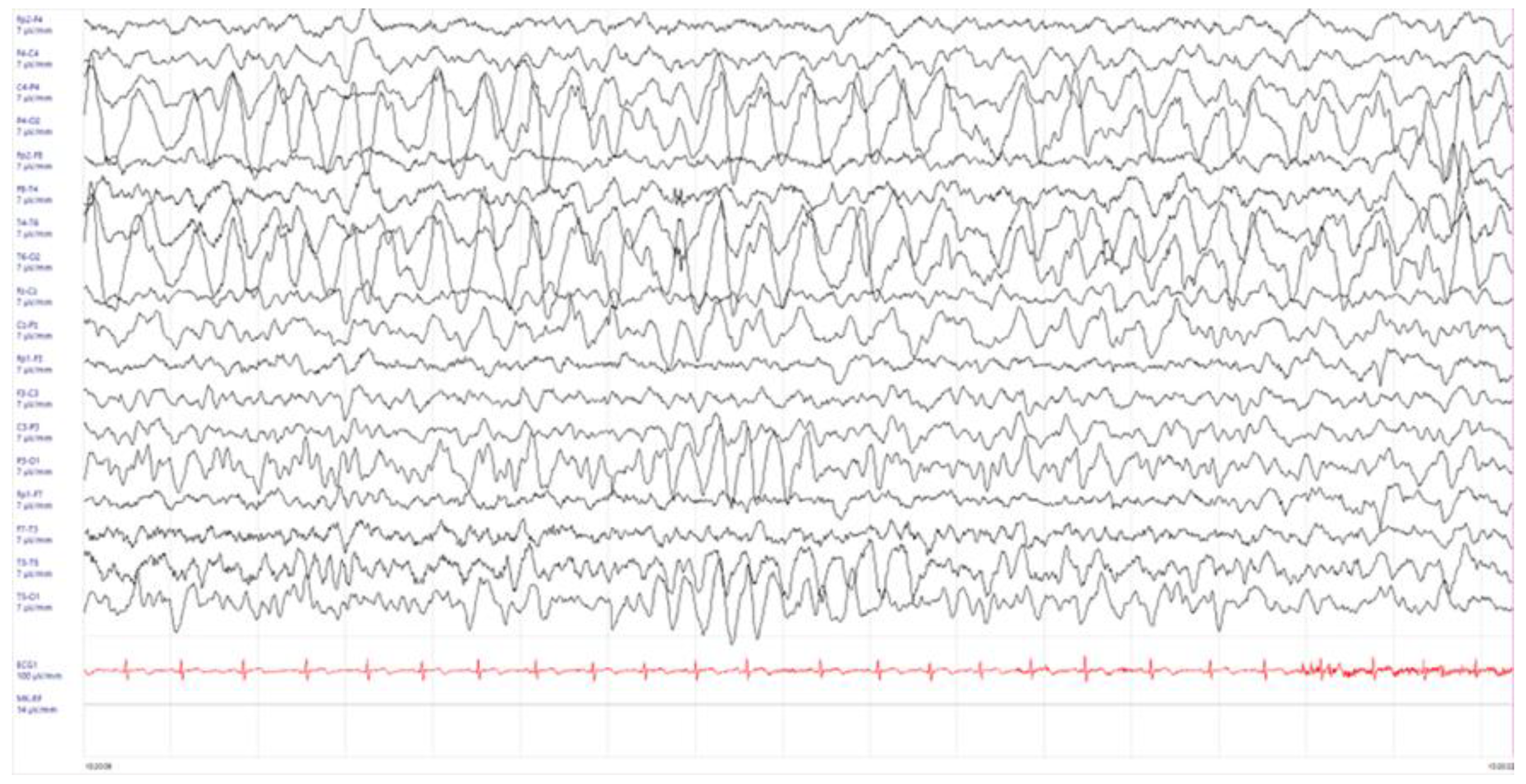
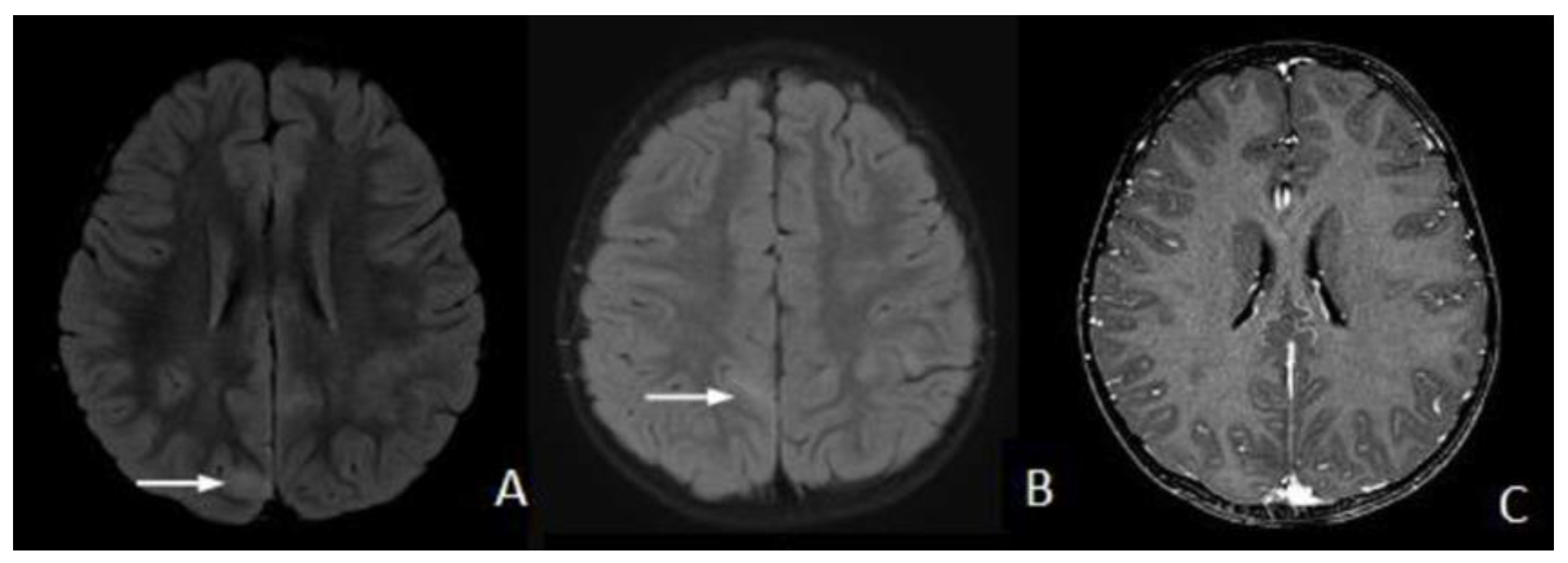
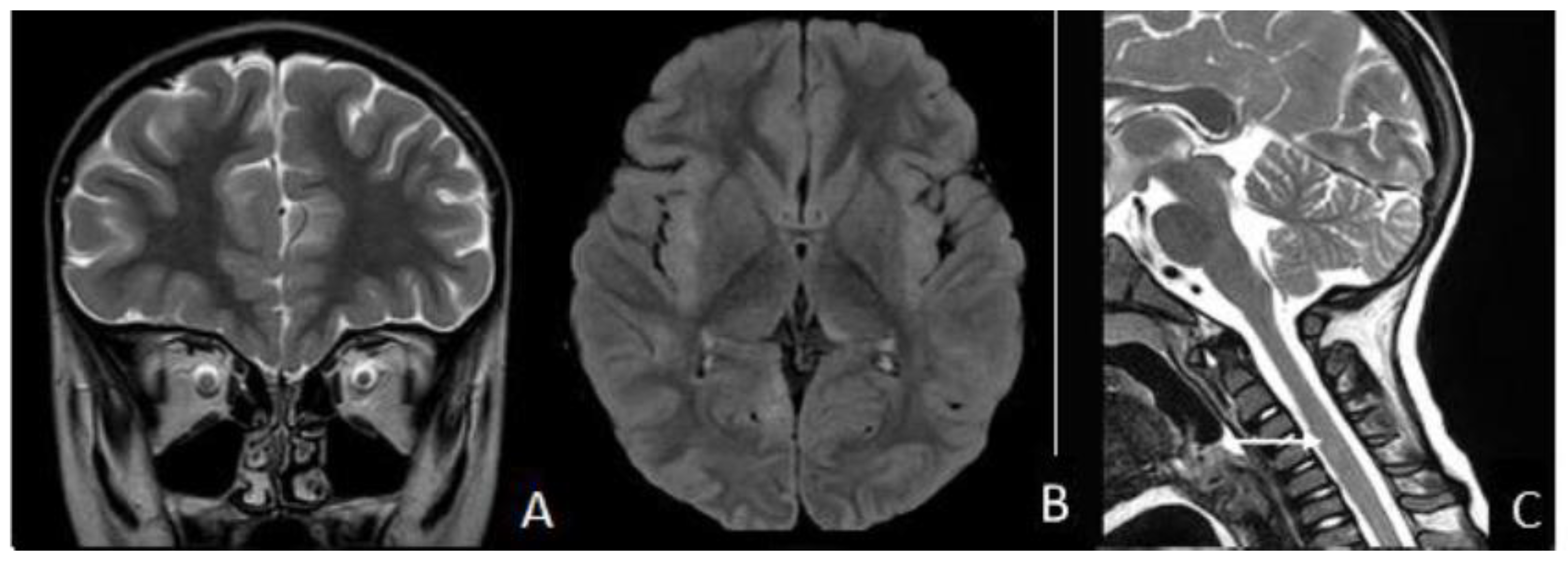
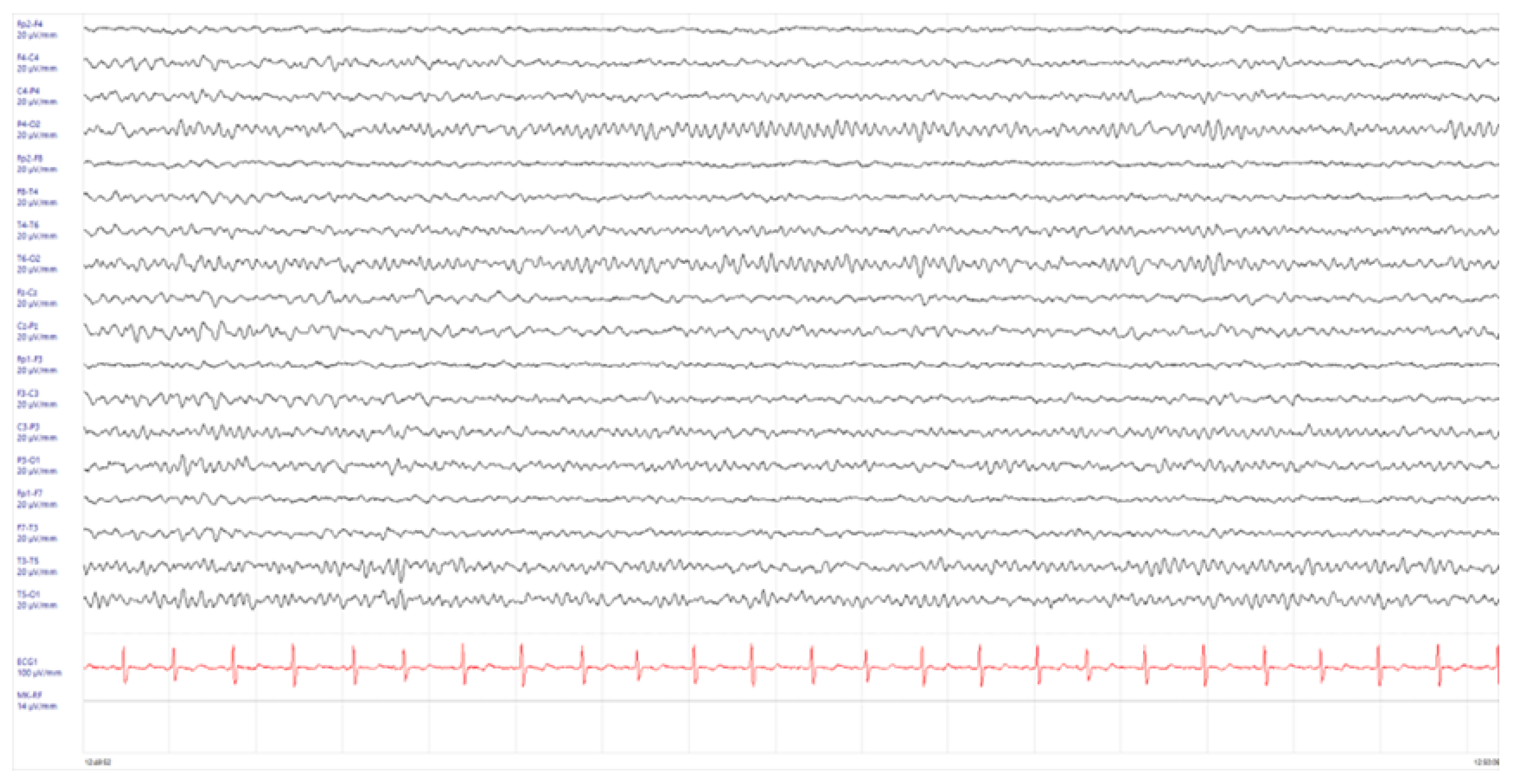
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esposito, S.; Principi, N.; Calabresi, P.; Rigante, D. An evolving redefinition of autoimmune encephalitis. Autoimmun. Rev. 2019, 18, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sechi, E.; Cacciaguerra, L.; Chen, J.J.; Mariotto, S.; Fadda, G.; Dinoto, A.; Lopez-Chiriboga, A.S.; Pittock, S.J.; Flanaga, E.P. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front. Neurol. 2022, 13, 885218. [Google Scholar] [CrossRef] [PubMed]
- Boulouis, G.; de Boysson, H.; Zuber, M.; Guillevin, L.; Meary, E.; Costalat, V.; Pagnoux, C.; Naggara, O. Primary Angiitis of the Central Nervous System: Magnetic Resonance Imaging Spectrum of Parenchymal, Meningeal, and Vascular Lesions at Baseline. Stroke 2017, 48, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, J.; Fan, Z.; Kang, J.; Wei, J.; Yin, X.; Yin, S. Case Report: Four Cases of Cortical/Brainstem Encephalitis Positive for Myelin Oligodendrocyte Glycoprotein Immunoglobulin G. Front. Neurol. 2022, 12, 775181. [Google Scholar] [CrossRef] [PubMed]
- Zhao-Fleming, H.H.; Valencia Sanchez, C.; Sechi, E.; Inbarasu, J.; Wijdicks, E.F.; Pittock, S.J.; Chen, J.J.; Wingerchuk, D.M.; Weinshenker, B.G.; Lopez-Chiriboga, S. CNS Demyelinating Attacks Requiring Ventilatory Support with Myelin Oligodendrocyte Glycoprotein or Aquaporin-4 Antibodies. Neurology 2021, 97, e1351–e1358. [Google Scholar] [PubMed]
- Duan, Y.; Zhuo, Z.; Li, H.; Tian, D.C.; Li, Y.; Yang, L.; Gao, C.; Zhang, T.; Zhang, X.; Shi, F.D. Brain structural alterations in MOG antibody diseases: A comparative study with AQP4 seropositive NMOSD and MS. J. Neurol. Neurosurg. Psychiatry 2021, 92, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, R.; Nakashima, I.; Takahashi, T.; Kaneko, K.; Akaishi, T.; Takai, Y.; Sato, D.K.; Nishiyama, S.; Misu, T.; Kuroda, H.; et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e322. [Google Scholar] [PubMed]
- Maeda, M.; Yagishita, A.; Yamamoto, T.; Sakuma, H.; Takeda, K. Abnormal hyperintensity within the subarachnoid space evaluated by fluid-attenuated inversion-recovery MR imaging:a spectrum of central nervous system diseases. Eur. Radiol. 2003, 13 (Suppl. S4), L192–L201. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Mirian, A.; Le, C.; Hosseini-Moghaddam, S.M.; Sharma, M.; Nicolle, M.W. Unilateral cortical FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis with Seizures (FLAMES): Characterization of a distinct clinico-radiographic syndrome. J. Neurol. 2019, 266, 2481–2487. [Google Scholar] [CrossRef]
- Foiadelli, T.; Gastaldi, M.; Scaranzin, S.; Franciotta, D.; Savasta, S. Seizures and myelin oligodendrocyte glycoprotein (MOG) antibodies: Two paradigmatic cases and a review of the literature. Mult. Scler. Relat. Disord. 2020, 41, 102011. [Google Scholar] [CrossRef]
- Jain, K.; Cherian, A.; Thomas, B. FLAMES: A novel burning entity in MOG IgG associated disease. Mult. Scler. Relat. Disord. 2021, 49, 102759. [Google Scholar] [CrossRef] [PubMed]
- Holay, Q.; Gazzola, S.; Quesnel, L.; Faivre, A. Migrating cortical lesion in FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures. J. Neurol. Neurosurg. Psychiatry 2023, 94, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Di Pietro, G.M.; Madini, B.; Mastrolia, M.V.; Rigante, D. A spectrum of inflammation and demyelination in acute disseminated encephalomyelitis (ADEM) of children. Autoimmun. Rev. 2015, 14, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Mohammad, S.; Tantsis, E.; Nguyen, T.K.; Merheb, V.; Fung, V.S.C.; White, O.B.; Broadley, S.; Lechner-Scott, J.; Vucic, S.; et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J. Neurol. Neurosurg. Psychiatry 2018, 89, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.X.; Yang, M.M.; Han, Y.J.; Gao, C.H.; Cao, J. FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures overlaying anti-N-methyl-D-aspartate receptor encephalitis: A case report and literature review. Front. Immunol. 2023, 14, 1149987. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.H.M.; Whittam, D.; Saviour, M.; Alorainy, A.; Mutch, K.; Linaker, S.; Solomon, T.; Bhojak, M.; Woodhall, M.; Waters, P.; et al. Seizures and Encephalitis in Myelin Oligodendrocyte Glycoprotein IgG Disease vs. Aquaporin 4 IgG Disease. JAMA Neurol. 2018, 75, 65–71. [Google Scholar] [CrossRef]
- Otani, T.; Irioka, T.; Igarashi, S.; Kaneko, K.; Takahashi, T.; Yokota, T. Self-remitting cerebral cortical encephalitis associated with myelin oligodendrocyte glycoprotein antibody mimicking acute viral encephalitis: A case report. Mult. Scler. Relat. Disord. 2020, 41, 102033. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, T.; Yamanaka, G.; Uryu, H.; Takeshita, M.; Morishita, N.; Morichi, S.; Ishida, Y.; Oana, S.; Terashi, H.; Shichino, H.; et al. Improvement in recurrent anti-myelin oligodendrocyte glycoprotein antibody—Positive cerebral cortical encephalitis not requiring anti—Inflammatory therapy following the decrease in cytokine/chemokine levels. Mult. Scler. Relat. Disord. 2020, 43, 102168. [Google Scholar] [CrossRef]
- Bruijstens, A.L.; Wendel, E.M.; Lechner, C.; Bartels, F.; Finke, C.; Breu, M.; Flet-Berliac, L.; de Chalus, A.; Adamsbaum, C.; Capobianco, M.; et al. E.U. paediatric MOG consortium consensus: Part 5—Treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur. J. Paediatr. Neurol. 2020, 29, 41–53. [Google Scholar] [CrossRef]
- Nosadini, M.; Eyre, M.; Giacomini, T.; Valeriani, M.; Della Corte, M.; Praticò, A.D.; Annovazzi, P.; Cordani, R.; Cordelli, D.M.; Crichiutti, G.; et al. Early Immunotherapy and Longer Corticosteroid Treatment Are Associated With Lower Risk of Relapsing Disease Course in Pediatric MOGAD. Neurol. Neuroimmunol. Neuroinflamm. 2022, 10, e200065. [Google Scholar] [CrossRef]
- Hacohen, Y.; Wong, Y.Y.; Lechner, C.; Jurynczyk, M.; Wright, S.; Konuskan, B.; Kalser, J.; Poulat, A.L.; Maurey, H.; Ganelin-Cohen, E.; et al. Disease Course and Treatment Responses in Children With Relapsing Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol. 2018, 75, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, E.; Kister, I. The Expanding Clinical Spectrum of Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Associated Disease in Children and Adults. Front. Neurol. 2020, 11, 960. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, L.; Mussi, N.; Grandinetti, R.; Turco, E.; Piccolo, B.; Ormitti, F.; Principi, N.; Esposito, S. FLAIR Hyperintense Cortical Lesions in a 4-Year-Old Child with Myelin Oligodendrocyte Glycoprotein (MOG)-Associated Encephalitis and Seizures: A Case Report. Children 2024, 11, 778. https://doi.org/10.3390/children11070778
Bernardi L, Mussi N, Grandinetti R, Turco E, Piccolo B, Ormitti F, Principi N, Esposito S. FLAIR Hyperintense Cortical Lesions in a 4-Year-Old Child with Myelin Oligodendrocyte Glycoprotein (MOG)-Associated Encephalitis and Seizures: A Case Report. Children. 2024; 11(7):778. https://doi.org/10.3390/children11070778
Chicago/Turabian StyleBernardi, Luca, Nicole Mussi, Roberto Grandinetti, Emanuela Turco, Benedetta Piccolo, Francesca Ormitti, Nicola Principi, and Susanna Esposito. 2024. "FLAIR Hyperintense Cortical Lesions in a 4-Year-Old Child with Myelin Oligodendrocyte Glycoprotein (MOG)-Associated Encephalitis and Seizures: A Case Report" Children 11, no. 7: 778. https://doi.org/10.3390/children11070778
APA StyleBernardi, L., Mussi, N., Grandinetti, R., Turco, E., Piccolo, B., Ormitti, F., Principi, N., & Esposito, S. (2024). FLAIR Hyperintense Cortical Lesions in a 4-Year-Old Child with Myelin Oligodendrocyte Glycoprotein (MOG)-Associated Encephalitis and Seizures: A Case Report. Children, 11(7), 778. https://doi.org/10.3390/children11070778