
Cephalometric Evaluation of Children with Short Stature of Genetic Etiology: A Review



Abstract
:1. Introduction
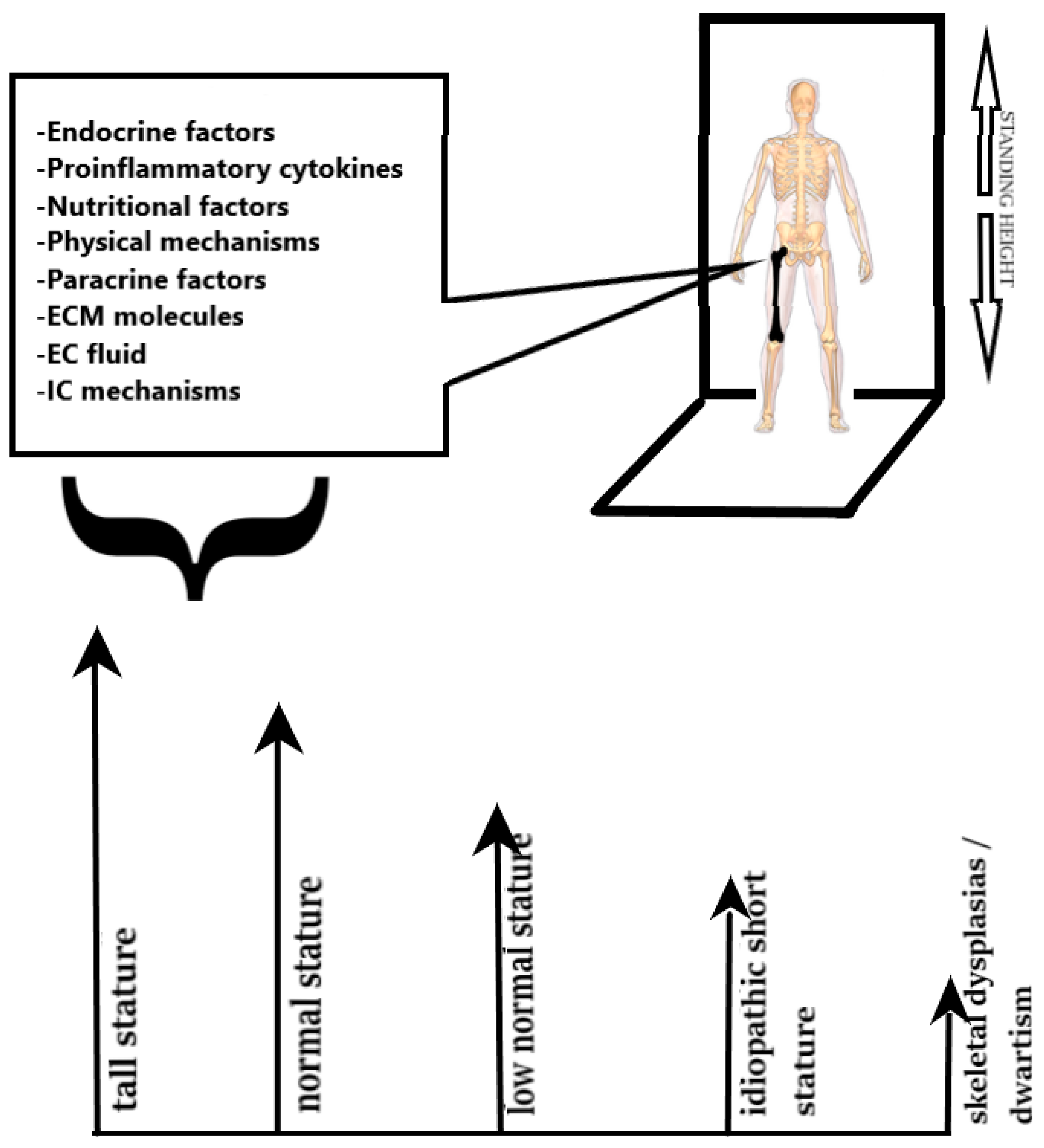
2. The Multifactorial Aetiology of Short Stature
3. Clinical Assessment of Children with Short Stature
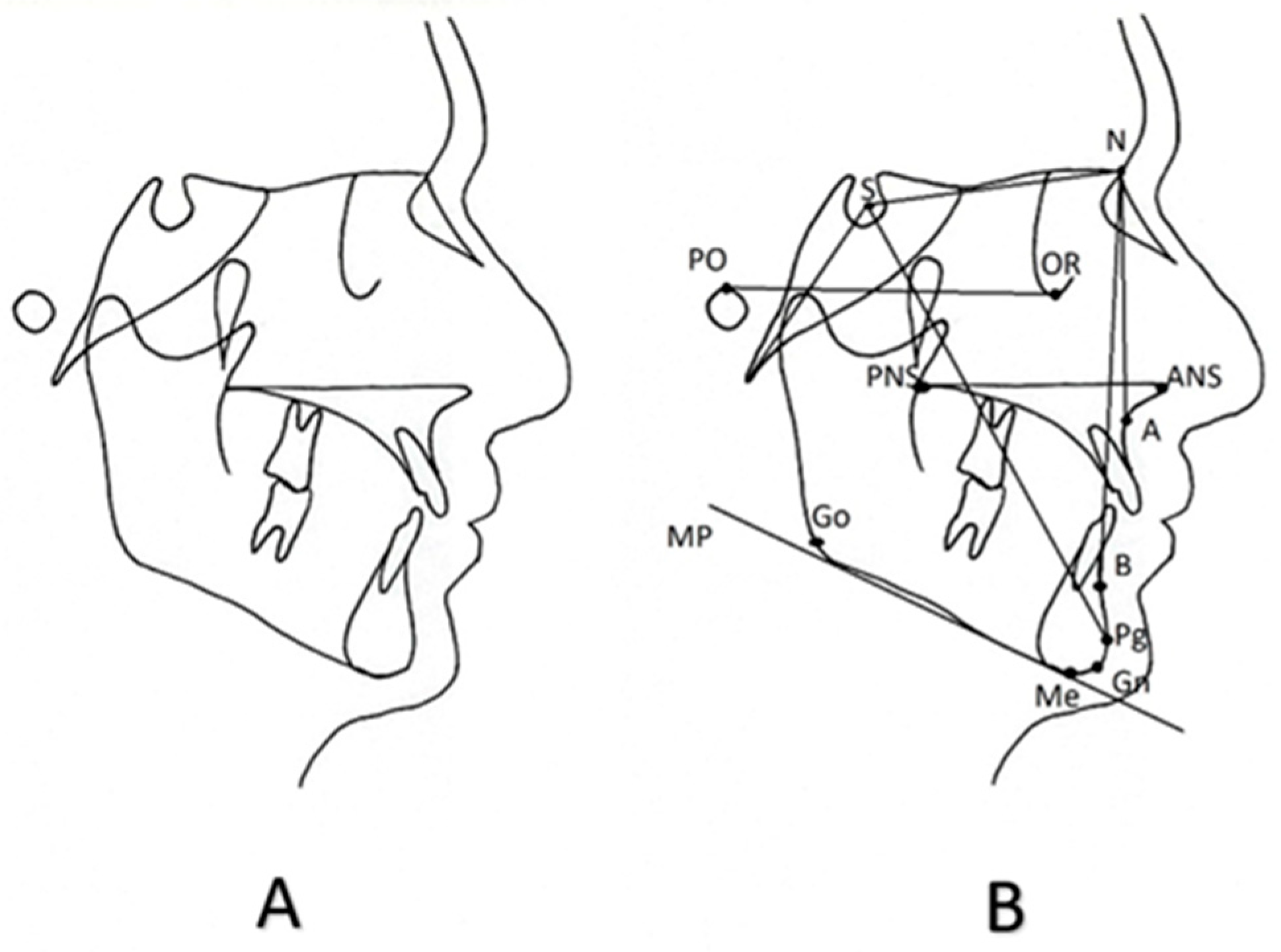
4. Cephalometric Assessment of Children with Short Stature
5. Cephalometric Data for Genetic Syndromes Associated with Short Stature
5.1. Achondroplasia (OMIM 100800)
5.2. Turner Syndrome (OMIM 309585)
5.3. Noonan Syndrome
5.4. Idiopathic Growth Hormone Deficiency (262400 Type IA; 612781 Type IB; 173100 Type II; 307200 Type III; 618157 Type IV)
5.5. Prader–Willi Syndrome (OMIM 176270)
5.6. Down Syndrome (OMIM 190685)
5.7. Muenke Syndrome (OMIM 602849)
5.8. Other Genetic Syndromes
6. Cephalometric Data for Gene Variants Associated with Short Stature
6.1. Growth Hormone Receptor Gene (GHR Gene)
6.2. Short Stature Homeobox Containing Gene (SHOX Gene)
6.3. Natriuretic Peptide Precursor C Gene (NPPC Gene)
6.4. Indian Hedgehog Gene (IHH Gene)
6.5. Fibroblast Growth Factor Receptor 3 Gene (FGFR3 Gene)
6.6. Parathyroid Hormone-like Hormone (PTHLH) or Parathyroid Hormone-Related Protein (PTHrP)
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wit, J.M.; Oostdijk, W.; Losekoot, M.; Van Duyvenvoorde, H.A.; Ruivenkamp, C.A.L.; Kant, S.G. MECHANISMS IN ENDOCRINOLOGY: Novel Genetic Causes of Short Stature. Eur. J. Endocrinol. 2016, 174, R145–R173. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, G.; Dimitropoulos, I.; Kourlaba, G.; Charmandari, E. The Effect of Treatment with Recombinant Human Growth Hormone (RhGH) on Linear Growth and Adult Height in Children with Idiopathic Short Stature (ISS): A Systematic Review and Meta-Analysis. J. Pediatr. Endocrinol. Metab. 2020, 33, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Rogol, A.D.; Hayden, G.F. Etiologies and Early Diagnosis of Short Stature and Growth Failure in Children and Adolescents. J. Pediatr. 2014, 164, S1–S14.e6. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Dattani, M.T.; Clayton, P.E. Controversies in the Diagnosis and Management of Growth Hormone Deficiency in Childhood and Adolescence. Arch. Dis. Child. 2016, 101, 96–100. [Google Scholar] [CrossRef]
- Grummer-Strawn, L.M.; Reinold, C.; Krebs, N.F.; Centers for Disease Control and Prevention (CDC). Use of World Health Organization and CDC Growth Charts for Children Aged 0–59 Months in the United States. MMWR Recomm. Rep. 2010, 59, 1–15. [Google Scholar]
- Rogol, A.D. Children with Asymptomatic Short Stature: What Is an Appropriate Evaluation? J. Pediatr. 2013, 163, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Height and Weight of Children of Hellenic Origin Aged 0–18 Years (2000–2001): Comparison with Data Collected during the Period 1978–1979 | Request PDF. Available online: https://www.researchgate.net/publication/284813940_Height_and_weight_of_children_of_Hellenic_origin_aged_0-18_years_2000-2001_Comparison_with_data_collected_during_the_period_1978-1979 (accessed on 30 April 2024).
- Wit, J.M.; Charmian Quigley, A.; Ranke, M.B. International Classification of Pediatric Endocrine Diagnoses. Horm. Res. Paediatr. 2016, 86, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Powls, A.; Botting, N.; Cooke, R.W.; Pilling, D.; Marlow, N. Growth Impairment in Very Low Birthweight Children at 12 Years: Correlation with Perinatal and Outcome Variables. Arch. Dis. Child. Fetal Neonatal Ed. 1996, 75, F152–F157. [Google Scholar] [CrossRef]
- Saunders, C.L.; Lejarraga, H.; Del Pino, M. Assessment of Head Size Adjusted for Height: An Anthropometric Tool for Clinical Use Based on Argentinian Data. Ann. Hum. Biol. 2006, 33, 415–423. [Google Scholar] [CrossRef]
- Ranke, M.B. Towards a Consensus on the Definition of Idiopathic Short Stature. Horm. Res. 1996, 45, 64–66. [Google Scholar] [CrossRef]
- Joseph, B. Short Stature and Altered Body Proportions. In Paediatric Orthopaedic Diagnosis; Springer: New Delhi, India, 2015; pp. 349–358. [Google Scholar] [CrossRef]
- Del Pino, M.; Orden, A.B.; Arenas, M.A.; Fano, V. Argentine References for the Assessment of Body Proportions from Birth to 17 Years of Age. Arch. Argent. Pediatr. 2017, 115, 234–240. [Google Scholar] [CrossRef]
- Duncan, W.J.; Fowler, R.S.; Farkas, L.G.; Ross, R.B.; Wright, A.W.; Bloom, K.R.; Huot, D.J.; Sondheimer, H.M.; Rowe, R.D.; Opitz, J.M. A Comprehensive Scoring System for Evaluating Noonan Syndrome. Am. J. Med. Genet. 1981, 10, 37–50. [Google Scholar] [CrossRef]
- Coi, A.; Santoro, M.; Garne, E.; Pierini, A.; Addor, M.C.; Alessandri, J.L.; Bergman, J.E.H.; Bianchi, F.; Boban, L.; Braz, P.; et al. Epidemiology of Achondroplasia: A Population-Based Study in Europe. Am. J. Med. Genet. A 2019, 179, 1791–1798. [Google Scholar] [CrossRef]
- Pineau, M.; Farrow, E.; Nicot, R.; Ferri, J. Achondroplasia: Orocraniofacial Features and Orthodontic-Surgical Management Guidelines Proposal. J. Craniofac. Surg. 2018, 29, 2186–2191. [Google Scholar] [CrossRef]
- Mm, C.; Gf, W.; Phillips, C. A Morphometric Analysis of the Craniofacial Configuration in Achondroplasia. J. Craniofac. Genet. Dev. Biol. Suppl. 1985, 1, 139–165. [Google Scholar]
- Celenk, P.; Arici, S.; Celenk, C. Oral Findings in a Typical Case of Achondroplasia. J. Int. Med. Res. 2003, 31, 236–238. [Google Scholar] [CrossRef]
- Shinohara, M.; Funakoshi, Y.; Takaishi, Y.; Hieda, T. A Case Report on the Achondroplasia and Its Dental Findings. Shoni Shikagaku Zasshi 1991, 29, 159–166. [Google Scholar]
- Bloom, M.W.; Murakami, S.; Cody, D.; Montufar-Solis, D.; Duke, P.J. Aspects of Achondroplasia in the Skulls of Dwarf Transgenic Mice: A Cephalometric Study. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 316–322. [Google Scholar] [CrossRef]
- Doswell, B.H.; Visootsak, J.; Brady, A.N.; Graham, J.M. Turner Syndrome: An Update and Review for the Primary Pediatrician. Clin. Pediatr. 2006, 45, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Tecuta-Busoi, A.-I.; Matei, M.; Florescu, L.M.; Gheonea, I.A. Developmental Abnormalities of the Skull Base in Patients with Turner Syndrome. Curr. Health Sci. J. 2020, 46, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Rizell, S. Dentofacial Morphology in Turner Syndrome Karyotypes. Swed. Dent. J. Suppl. 2012, 225, 7–98. [Google Scholar]
- Rizell, S.; Barrenas, M.-L.; Andlin-Sobocki, A.; Stecksen-Blicks, C.; Kjellberg, H. 45,X/46,XX Karyotype Mitigates the Aberrant Craniofacial Morphology in Turner Syndrome. Eur. J. Orthod. 2013, 35, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Perkiomaki, M.R.; Kyrkanides, S.; Niinimaa, A.; Alvesalo, L. The Relationship of Distinct Craniofacial Features between Turner Syndrome Females and Their Parents. Eur. J. Orthod. 2005, 27, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Eklund, M.; Kotilainen, J.; Evälahti, M.; Waltimo-Sirén, J. Cephalometric Analysis of Pharyngeal Airway Space Dimensions in Turner Syndrome. Eur. J. Orthod. 2012, 34, 219–225. [Google Scholar] [CrossRef]
- Dumancic, J.; Kaic, Z.; Varga, M.L.; Lauc, T.; Dumic, M.; Milosevic, S.A.; Brkic, H. Characteristics of the Craniofacial Complex in Turner Syndrome. Arch. Oral Biol. 2010, 55, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.; Sonnesen, L.; Kjaer, M.S.; Fischer Hansen, B.; Kjær, I. The Prenatal Cranial Base Complex and Hand in Turner Syndrome. Eur. J. Orthod. 2000, 22, 185–194. [Google Scholar] [CrossRef]
- Simmons, K.E. Growth Hormone and Craniofacial Changes: Preliminary Data from Studies in Turner’s Syndrome. Pediatrics 1999, 104, 1021–1024. [Google Scholar] [CrossRef]
- Hass, A.D.; Simmons, K.E.; Davenport, M.L.; Proffit, W.R. The Effect of Growth Hormone on Craniofacial Growth and Dental Maturation in Turner Syndrome. Angle Orthod. 2001, 71, 50–59. [Google Scholar] [CrossRef]
- Corvo, G.; Tartaro, G.P.; Stoppoloni, F.; Balzano, G. Cephalometric Evaluation of Patients with Turner Syndrome. Authors’ Experience. Minerva Stomatol. 1998, 47, 127–133. [Google Scholar]
- Rongen-Westerlaken, C.; vd Born, E.; Prahl-Andersen, B.; Rikken, B.; Teunenbroek, V.; Kamminga, N.; vd Tweel, I.; Otten, B.J.; Delamarre vd Waal, H.A.; Drayer, N.M. Shape of the Craniofacial Complex in Children with Turner Syndrome. J. Biol. Buccale 1992, 20, 185–190. [Google Scholar]
- Rongen-Westerlaken, C.; vd Born, E.; Prahl-Andersen, B.; v Teunenbroek, A.; Manesse, P.; Otten, B.; vd Tweel, I.; Kuijpers-Jagtman, A.; Delemarre vd Waal, H.; Drayer, N.; et al. Effect of Growth Hormone Treatment on Craniofacial Growth in Turner’s Syndrome. Acta Paediatr. 1993, 82, 364–368. [Google Scholar] [CrossRef]
- Midtbo, M.; Wisth, P.J.; Halse, A. Craniofacial Morphology in Young Patients with Turner Syndrome. Eur. J. Orthod. 1996, 18, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Peltomäki, T.; Alvesalo, L.; Isotupa, K. Shape of the Craniofacial Complex in 45,X Females: Cephalometric Study. J. Craniofac. Genet. Dev. Biol. 1989, 9, 331–338. [Google Scholar] [PubMed]
- Jensen, B.L. Craniofacial Morphology in Turner Syndrome. J. Craniofac. Genet. Dev. Biol. 1985, 5, 327–340. [Google Scholar] [PubMed]
- Rzymski, K.; Kosowicz, J. Abnormal Basal Angle of the Skull in Sex Chromosome Aberrations. Acta Radiol. Diagn. 1976, 17, 669–675. [Google Scholar] [CrossRef]
- Rzymski, K.; Kosowicz, J. The Skull in Gonadal Dysgenesis a Roentgenometric Study. Clin. Radiol. 1975, 26, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Tai, K.; Sato, Y. Management of Klippel-Feil Syndrome Combined with Turner Syndrome: A Case Report. Int. J. Orthod. Milwaukee 2013, 24, 37–42. [Google Scholar] [PubMed]
- Horowitz, S.L.; Morishima, A.; Vinkka, H. The Position of the External Ear in Turner’s Syndrome. Clin. Genet. 1976, 9, 333–340. [Google Scholar] [CrossRef]
- Weiss, E.; Loevy, H.; Saunders, A.; Pruzansky, S.; Rosenthal, I.M.; Opitz, J.M. Monozygotic Twins Discordant for Ullrich-Turner Syndrome. Am. J. Med. Genet. 1982, 13, 389–399. [Google Scholar] [CrossRef]
- Filipsson, R.; Lindsten, J.; Almqvist, S. Time of Eruption of The Permanent Teeth, Cephalometric and Tooth Measurement and Sulphation Factor Activity in 45 Patients with Turner’s Syndrome with Different Types of X Chromosome Aberrations. Acta Endocrinol. 1965, 48, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Ahiko, N.; Baba, Y.; Tsuji, M.; Horikawa, R.; Moriyama, K. Investigation of Maxillofacial Morphology and Oral Characteristics with Turner Syndrome and Early Mixed Dentition. Congenit. Anom. 2019, 59, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Cazzolla, A.P.; Lo Muzio, L.; Di Fede, O.; Lacarbonara, V.; Colaprico, A.; Testa, N.F.; Giuseppe, T.; Zhurakivska, K.; Marzo, G.; Lacaita, M.G. Orthopedic-orthodontic Treatment of the Patient with Turner’s Syndrome: Review of the Literature and Case Report. Spec. Care Dent. 2018, 38, 239–248. [Google Scholar] [CrossRef]
- Aristizábal, J.F.; Smit, R.M. Two-Phase Orthodontic Treatment in a Patient with Turner Syndrome: An Unusual Case of Deep Bite. Cleft Palate-Craniofac. J. 2015, 52, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.A. Orthodontic Treatment for Patients with Turner Syndrome. Am. J. Orthod. Dentofac. Orthop. 2001, 120, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Ogiuchi, H.; Takano, K.; Tanaka, M.; Hizuka, N.; Takagi, S.; Sangu, Y.; Shizume, K.; Kawanishi, I. Oro-Maxillofacial Development in Patients with Turner’s Syndrome. Endocrinol. Jpn. 1985, 32, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Svanberg, C.; Norevall, L.-I.; Ekman, B.; Wahlberg, J.; Bågesund, M. Cephalometric Analysis of Adults with Turner Syndrome. Swed. Dent. J. 2016, 40, 33–41. [Google Scholar]
- Takeyama, H.; Honzawa, O.; Hozaki, T.; Kiyomura, H. A Case of Open Bite with Turner’s Syndrome. Am. J. Orthod. Dentofac. Orthop. 1990, 97, 505–509. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Redman, R.S.; Shapiro, B.L. Effect of X-Chromosome Aneuploidy on Jaw Growth. J. Dent. Res. 1965, 44, 269–282. [Google Scholar] [CrossRef]
- Bagattoni, A.; Lardani, L.; Vanni, A.; Costi, T. Craniofacial and Occlusal Features of Individuals with Turner Syndrome: A Cephalometric Study. J. Biol. Regul. Homeost. Agents 2021, 35, 95–106. [Google Scholar] [CrossRef]
- Juloski, J.; Dumančić, J.; Šćepan, I.; Lauc, T.; Milašin, J.; Kaić, Z.; Dumić, M.; Babić, M. Growth Hormone Positive Effects on Craniofacial Complex in Turner Syndrome. Arch. Oral Biol. 2016, 71, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Juloski, J.; Glisic, B.; Scepan, I.; Milasin, J.; Mitrovic, K.; Babic, M. Ontogenetic Changes of Craniofacial Complex in Turner Syndrome Patients Treated with Growth Hormone. Clin. Oral Investig. 2013, 17, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Babic, M.; Glisic, B.; Scepan, I. Mandibular Growth Pattern in Turner’s Syndrome. Eur. J. Orthod. 1997, 19, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Perkiomaki, M.R.; Alvesalo, L. Palatine Ridges and Tongue Position in Turner Syndrome Subjects. Eur. J. Orthod. 2008, 30, 163–168. [Google Scholar] [CrossRef]
- Bhambhani, V.; Muenke, M. Noonan Syndrome. Am. Fam. Physician 2014, 89, 37–43. [Google Scholar]
- Miyamoto, J.J.; Yabunaka, T.; Moriyama, K. Cervical Characteristics of Noonan Syndrome. Eur. J. Orthod. 2014, 36, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B.; Heidemann, P.; Knupfer, C.; Enders, H.; Schmaltz, A.A.; Bierich, J.R. Noonan Syndrome: Growth and Clinical Manifestations in 144 Cases. Eur. J. Pediatr. 1988, 148, 220–227. [Google Scholar] [CrossRef]
- Chatzistavrou, E.; Andreadis, G. Noonan Syndrome in 12-Year-Old Male: Case Report and Orthodontic Management of the Occlusion. Balk. J. Dent. Med. 2020, 24, 118–126. [Google Scholar] [CrossRef]
- Kawakami, M.; Yamamoto, K.; Shimomura, T.; Kirita, T. Surgical Orthodontic Treatment for Open Bite in Noonan Syndrome Patient: A Case Report. Cleft Palate-Craniofac. J. 2016, 53, 253–258. [Google Scholar] [CrossRef]
- Emral, M.E.; Akcam, M.O. Noonan Syndrome: A Case Report. J. Oral Sci. 2009, 51, 301–306. [Google Scholar] [CrossRef]
- Bagattoni, S.; Costi, T.; D’Alessandro, G.; Toni, S.; Gatto, M.R.; Piana, G. Craniofacial and Occlusal Features of Children with Noonan Syndrome. Am. J. Med. Genet. A 2021, 185, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Cardiel Ríos, S.A. Correction of a Severe Class II Malocclusion in a Patient with Noonan Syndrome. Am. J. Orthod. Dentofac. Orthop. 2016, 150, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Al Qabbani, H. Noonan Syndrome: A Case Report. Int. J. Oral Health Dent. 2017, 3, 238–239. [Google Scholar] [CrossRef]
- Okada, M.; Sasaki, N.; Kaihara, Y.; Okada, R.; Amano, H.; Miura, K.; Kozai, K. Oral Findings in Noonan Syndrome: Report of a Case. J. Oral Sci. 2003, 45, 117–121. [Google Scholar] [CrossRef]
- Oliveira-Neto, L.A.; Melo, M.F.B.; Franco, A.A.; Oliveira, A.H.A.; Souza, A.H.O.; Valença, E.H.O.; Britto, I.M.P.A.; Salvatori, R.; Aguiar-Oliveira, M.H. Cephalometric Features in Isolated Growth Hormone Deficiency. Angle Orthod. 2011, 81, 578–583. [Google Scholar] [CrossRef]
- Kjellberg, H.; Wikland, K.A. A Longitudinal Study of Craniofacial Growth in Idiopathic Short Stature and Growth Hormone-Deficient Boys Treated with Growth Hormone. Eur. J. Orthod. 2007, 29, 243–250. [Google Scholar] [CrossRef]
- Funatsu, M.; Sato, K.; Mitani, H. Effects of Growth Hormone on Craniofacial Growth. Angle Orthod. 2006, 76, 970–977. [Google Scholar] [CrossRef]
- Vasconcelos, G.; Stenehjem, J.S.; Axelsson, S.; Saeves, R. Craniofacial and Dentoalveolar Morphology in Individuals with Prader–Willi Syndrome: A Case-Control Study. Orphanet J. Rare Dis. 2022, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Giuca, M.R.; Inglese, R.; Caruso, S.; Gatto, R.; Marzo, G.; Pasini, M. Craniofacial Morphology in Pediatric Patients with Prader–Willi Syndrome: A Retrospective Study. Orthod. Craniofac. Res. 2016, 19, 216–221. [Google Scholar] [CrossRef]
- Schaedel, R.; Poole, A.E.; Cassidy, S.B. Cephalometric Analysis of the Prader-Willi Syndrome. Am. J. Med. Genet. 1990, 36, 484–487. [Google Scholar] [CrossRef]
- Gopinath, T.; Ganesh, S.; Subramani, V. Role of Facial Index and Odontometric Parameters in the Establishment of Stature and Gender of Individuals. J. Pharm. Bioallied Sci. 2021, 13, S1068–S1073. [Google Scholar] [CrossRef] [PubMed]
- Jesuino, F.A.S.; Valladares-Neto, J. Craniofacial Morphological Differences between Down Syndrome and Maxillary Deficiency Children. Eur. J. Orthod. 2013, 35, 124–130. [Google Scholar] [CrossRef] [PubMed]
- van Marrewijk, D.J.F.; van Stiphout, M.A.E.; Reuland-Bosma, W.; Bronkhorst, E.M.; Ongkosuwito, E.M. The Relationship between Craniofacial Development and Hypodontia in Patients with Down Syndrome. Eur. J. Orthod. 2016, 38, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, J.; Biedma, B.; Rodríguez, M.; Mora, M.; Cunqueiro, M.; Pazos, M. Cephalometrics in Children with Down’s Syndrome. Pediatr. Radiol. 2002, 32, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Faruqui, S.; Fida, M.; Shaikh, A. Cervical Vertebral Anomalies in Skeletal Malocclusions: A Cross-Sectional Study on Orthodontic Patients at the Aga Khan University Hospital, Pakistan. Indian J. Dent. Res. 2014, 25, 480. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, E.B.; Wu, J.K.; Sullivan, S.R.; Vasudavan, S.; Padwa, B.L.; Rogers, G.F.; Mulliken, J.B. Craniofacial Growth in Patients with FGFR3Pro250Arg Mutation after Fronto-Orbital Advancement in Infancy. J. Craniofac. Surg. 2011, 22, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, F.K.; Mui, B.W.H.; Almpani, K.; Jani, P.; Keyvanfar, C.; Iqbal, K.; Paravastu, S.S.; Arora, D.; Orzechowski, P.; Merling, R.K.; et al. Quantitative Craniofacial Analysis and Generation of Human Induced Pluripotent Stem Cells for Muenke Syndrome: A Case Report. J. Dev. Biol. 2021, 9, 39. [Google Scholar] [CrossRef]
- Reinhart, E.; Eulert, S.; Bill, J.; Würzler, K.; Phan The, L.; Reuther, J. Typische Merkmale Des Kraniofazialen Wachstums Beim FGFR3-Assoziierten Koronarnahtsynostosesyndrom (so Genannte Muenke-Kraniosynostose). Mund- Kiefer- Gesichtschirurgie 2003, 7, 132–137. [Google Scholar] [CrossRef]
- Muenke, M.; Gripp, K.W.; McDonald-McGinn, D.M.; Gaudenz, K.; Whitaker, L.A.; Bartlett, S.P.; Markowitz, R.I.; Robin, N.H.; Nwokoro, N.; Mulvihill, J.J.; et al. A Unique Point Mutation in the Fibroblast Growth Factor Receptor 3 Gene (FGFR3) Defines a New Craniosynostosis Syndrome. Am. J. Hum. Genet. 1997, 60, 555–564. [Google Scholar]
- Samra, F.; Bauder, A.R.; Swanson, J.W.; Whitaker, L.A.; Bartlett, S.P.; Taylor, J.A. Assessing the Midface in Muenke Syndrome: A Cephalometric Analysis and Review of the Literature. J. Plast. Reconstr. Aesthetic Surg. 2016, 69, 1285–1290. [Google Scholar] [CrossRef]
- Arnaud, E.; Meneses, P.; Lajeunie, E.; Thorne, J.A.; Marchac, D.; Renier, D. Postoperative Mental and Morphological Outcome for Nonsyndromic Brachycephaly. Plast. Reconstr. Surg. 2002, 110, 6–12. [Google Scholar] [CrossRef]
- Lewyllie, A.; Roosenboom, J.; Indencleef, K.; Claes, P.; Swillen, A.; Devriendt, K.; Carels, C.; Cadenas De Llano-Pérula, M.; Willems, G.; Hens, G.; et al. A Comprehensive Craniofacial Study of 22q11.2 Deletion Syndrome. J. Dent. Res. 2017, 96, 1386–1391. [Google Scholar] [CrossRef]
- Lia, E.N.; Otero, S.A.M.; Ferraz, M.; Gonçalves, L.P.V. Oral Aspects of 49, XXXXY Syndrome: A Case Report. J. Dent. Child 2007, 74, 136–139. [Google Scholar]
- Wulfsberg, E.A.; Campbell, A.B.; Lurie, I.W.; Eanet, K.R. Confirmation of the Catania Brachydactylous Type of Acrofacial Dysostosis: Report of a Second Family. Am. J. Med. Genet. 1996, 63, 554–557. [Google Scholar] [CrossRef]
- Defabianis, P.; Mussa, A.; Ninivaggi, R.; Carli, D.; Romano, F. Maxillo-Facial Morphology in Beckwith-Wiedemann Syndrome: A Preliminary Study on (Epi)Genotype-Phenotype Association in Caucasians. Int. J. Environ. Res. Public Health 2022, 19, 2448. [Google Scholar] [CrossRef]
- Nojima, K.; Onoda, M.; Nishii, Y.; Sueishi, K. Orthodontic Treatment for Bloch-Sulzberger Syndrome in Patient with Cleft Lip and Palate. Bull. Tokyo Dent. Coll. 2017, 58, 259–267. [Google Scholar] [CrossRef]
- Bender, C.V.; da Silveira, H.L.D.; dos Santos, N.S.; Cavagni, J.; Rados, P.V.; John, A.B.; De Souza, C.F.M.; Giugliani, R.; Visioli, F. Oral, Dental, and Craniofacial Features in Chronic Acid Sphingomyelinase Deficiency. Am. J. Med. Genet. A 2020, 182, 2891–2901. [Google Scholar] [CrossRef]
- Bloch-Zupan, A.; Rousseaux, M.; Lauge, V.; Schmittbuhl, M.; Mathis, R.; Desforges, E.; Koob, M.; Zaloszyc, A.; Dollfus, H.; Laugel, V. A Possible Cranio-Oro-Facial Phenotype in Cockayne Syndrome. Orphanet J. Rare Dis. 2013, 8, 9. [Google Scholar] [CrossRef]
- Yapijakis, C.; Pachis, N.; Sotiriadou, T.; Vaila, C.; Michopoulou, V.; Vassiliou, S. Molecular Mechanisms Involved in Craniosynostosis. In Vivo 2023, 37, 36–46. [Google Scholar] [CrossRef]
- Yang, I.-H.; Chung, J.H.; Cho, I.-S.; Kim, S.; Baek, S.-H. Effect of Early Spheno-Occipital Synchondrosis Fusion in Preadolescent Patients with Syndromic Craniosynostosis on Craniofacial Skeletal Patterns: A Preliminary Study Using Cephalometric Analysis. J. Craniofac. Surg. 2022, 33, 179–182. [Google Scholar] [CrossRef]
- Wang, H.; Hung, K.; Zhao, K.; Wang, Y.; Wang, F.; Wu, Y. Anatomical Analysis of Zygomatic Bone in Ectodermal Dysplasia Patients with Oligodontia. Clin. Implant. Dent. Relat. Res. 2019, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Baba, Y.; Tsuji, M.; Fukuoka, H.; Ogawa, T.; Ohkuma, M.; Moriyama, K. Dentomaxillofacial Characteristics of Ectodermal Dysplasia. Congenit. Anom. 2015, 55, 42–48. [Google Scholar] [CrossRef]
- Hart, T.C.; Kyrkanides, S. Cephalometric Analysis of Rapp-Hodgkin Syndrome. J. Med. Genet. 1994, 31, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Tuna, E.B.; Koruyucu, M.; Kürklü, E.; Çifter, M.; Gençay, K.; Seymen, F.; Tüysüz, B. Oral and Craniofacial Manifestations of Ellis–van Creveld Syndrome: Case Series. J. Cranio-Maxillofac. Surg. 2016, 44, 919–924. [Google Scholar] [CrossRef]
- Danaei, S.M.; Karamifar, A.; Sardarian, A.; Shahidi, S.; Karamifar, H.; Alipour, A.; Ghodsi Boushehri, S. Measuring Agreement between Cervical Vertebrae and Hand-Wrist Maturation in Determining Skeletal Age: Reassessing the Theory in Patients with Short Stature. Am. J. Orthod. Dentofac. Orthop. 2014, 146, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou-Kyrkanidou, E.; Vrahopoulos, T.P.; Eliades, G.; Vastardis, H.; Tosios, K.; Vrotsos, I.A. Periodontitis Associated with Hajdu-Cheney Syndrome. J. Periodontol. 2007, 78, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Bahar Tuna, E.; Sulun, T.; Rosti, O.; El Abdallah, F.; Kayserili, H.; Aktoren, O. Craniodentofacial Manifestations in Hallermann-Streiff Syndrome. J. Craniomandib. Sleep Pract. 2009, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tokura, T.; Miyazaki, A.; Igarashi, T.; Dehari, H.; Kobayashi, J.; Miki, Y.; Ogi, K.; Sonoda, T.; Yotsuyanagi, T.; Hiratsuka, H. Quantitative Evaluation of Cephalometric Radiographs of Patients with Hemifacial Microsomia. Cleft Palate-Craniofac. J. 2019, 56, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Tuna, E.; Marşan, G.; Gençay, K.; Seymen, F. Craniofacial and Dental Characteristics of Kabuki Syndrome: Nine Years Cephalometric Follow-up. J. Clin. Pediatr. Dent. 2012, 36, 393–400. [Google Scholar] [CrossRef]
- Ozdiler, E.; Akcam, M.O.; Sayin, M.O. Craniofacial Characteristics of Klippel-Feil Syndrome in an Eight Year Old Female. J. Clin. Pediatr. Dent. 2000, 24, 249–254. [Google Scholar]
- Katge, F.A.; Rusawat, B.D.; Shivasharan, P.R.; Patil, D.P. Langer-Giedion Syndrome: A Rare Case Report. J. Dent. 2016, 17, 238–241. [Google Scholar]
- Yasunaga, M.; Ishikawa, H.; Yanagita, K.; Tamaoki, S. An Orthodontic Perspective on Larsen Syndrome. BMC Oral Health 2021, 21, 111. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Tagariello, T.; Piana, G. Oral and Craniofacial Findings in a Patient with Methylmalonic Aciduria and Homocystinuria: Review and a Case Report. Minerva Stomatol. 2010, 59, 129–137. [Google Scholar] [PubMed]
- Telich-Tarriba, J.E.; Amador-Lara, A.; Quiroz-Barrios, J.; Cardenas-Mejia, A. Cephalometric Analysis of the Craniofacial Morphology in Patients with Moebius Syndrome. J. Craniofac. Surg. 2021, 32, 2446–2448. [Google Scholar] [CrossRef] [PubMed]
- Fontinha, C.; Engvall, M.; Sjögreen, L.; Kiliaridis, S. Craniofacial Morphology and Growth in Young Patients with Congenital or Childhood Onset Myotonic Dystrophy. Eur. J. Orthod. 2018, 40, 544–548. [Google Scholar] [CrossRef]
- Romero, M.; Franco, B.; del Pozo, J.S.; Romance, A. Buccal Anomalies, Cephalometric Analysis and Genetic Study of Two Sisters with Orofaciodigital Syndrome Type I. Cleft Palate-Craniofac. J. 2007, 44, 660–666. [Google Scholar] [CrossRef]
- Martins, F.; Roussen, A.C.; Rezende, N.; Hiraoka, C.; Zamunaro, M.; Gallottini, M. Oral and Cephalometric Study in Brazilian Rubinstein–Taybi Syndrome Patients. Spec. Care Dent. 2022, 42, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kjær, I.; Hansen, N.; Becktor, K.B.; Birkebæk, N.; Balslev, T. Craniofacial Morphology, Dentition, and Skeletal Maturity in Four Siblings with Seckel Syndrome. Cleft Palate-Craniofac. J. 2001, 38, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Vo Quang, S.; Galliani, E.; Eche, S.; Tomat, C.; Fauroux, B.; Picard, A.; Kadlub, N. Contribution of a Better Maxillofacial Phenotype in Silver–Russell Syndrome to Define a Better Orthodontics and Surgical Management. J. Stomatol. Oral Maxillofac. Surg. 2019, 120, 110–115. [Google Scholar] [CrossRef]
- Taniyama, T.; Kitai, N.; Iguchi, Y.; Murakami, S.; Yanagi, M.; Takada, K. Craniofacial Morphology in a Patient with Simpson-Golabi-Behmel Syndrome. Cleft Palate-Craniofac. J. 2003, 40, 550–555. [Google Scholar] [CrossRef]
- Bayram, M.; Yildirim, M.; Seymen, F. Clinical and Oral Findings of a Patient with Simpson–Golabi–Behmel Syndrome. Eur. Arch. Paediatr. Dent. 2015, 16, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Nota, A.; Ehsani, S.; Pittari, L.; Gastaldi, G.; Tecco, S. Rare Case of Skeletal Third Class in a Subject Suffering from Solitary Median Maxillary Central Incisor Syndrome (SMMCI) Associated to Panhypopituitarism. Head Face Med. 2021, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, H.; Shetye, P.R.; Grayson, B.H.; McCarthy, J.G. Cephalometric Assessment of Craniofacial Morphology in Patients with Treacher Collins Syndrome. J. Craniofac. Surg. 2013, 24, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Belostoky, L. Craniofacial Morphology of Children with Williams Syndrome. Cleft Palate Craniofac. J. 1993, 30, 343–349. [Google Scholar] [CrossRef]
- Danneels, F.; Verdonck, A.; Indencleef, K.; Declerck, D.; Willems, G.; Cadenas De Llano-Pérula, M. Determination of Craniofacial and Dental Characteristics of Individuals with Williams-Beuren Syndrome by Using 3D Facial Scans and Radiographs. Orthod. Craniofac. Res. 2022, 25, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Li, Y.; Shao, Q.; Zhang, C.; Kou, S.; Yang, W.; Zhang, M.; Ban, B. Clinical and Genetic Evaluation of Children with Short Stature of Unknown Origin. BMC Med. Genom. 2023, 16, 194. [Google Scholar] [CrossRef]
- Tobón-Arroyave, S.I.; Jiménez-Arbeláez, G.A.; Alvarado-Gómez, V.A.; Isaza-Guzmán, D.M.; Flórez-Moreno, G.A.; Pérez-Cano, M.I. Association Analysis between Rs6184 and Rs6180 Polymorphisms of Growth Hormone Receptor Gene Regarding Skeletal-Facial Profile in a Colombian Population. Eur. J. Orthod. 2018, 40, 378–386. [Google Scholar] [CrossRef]
- Depeyre, A.; Schlund, M.; Nicot, R.; Ferri, J. Dental and Maxillofacial Signs in Leri-Weill Dyschondrosteosis. J. Oral Maxillofac. Surg. 2019, 77, 762–768. [Google Scholar] [CrossRef]
- Al Kaissi, A.; Shboul, M.; Kenis, V.; Grill, F.; Ganger, R.; Kircher, S.G. Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan. Medicines 2019, 6, 60. [Google Scholar] [CrossRef]
- De Leenheer, E.M.R.; Kuijpers-Jagtman, A.M.; Sengers, R.C.A.; Oudesluijs, G.G.; Rappold, G.A.; Cremers, C.W.R.J. Congenital Conductive Hearing Loss in Dyschondrosteosis. Ann. Otol. Rhinol. Laryngol. 2003, 112, 153–158. [Google Scholar] [CrossRef]
- Nakao, K.; Okubo, Y.; Yasoda, A.; Koyama, N.; Osawa, K.; Isobe, Y.; Kondo, E.; Fujii, T.; Miura, M.; Nakao, K.; et al. The Effects of C-Type Natriuretic Peptide on Craniofacial Skeletogenesis. J. Dent. Res. 2013, 92, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Rabie, A.B.M.; Hägg, U. Indian Hedgehog: A Mechanotransduction Mediator in Condylar Cartilage. J. Dent. Res. 2004, 83, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanya, R.; Prasad, S.; Prasad, R.; Mogra, S.; Shetty, V.; Rao, V. Mutations in FGFR3 Gene Associated with Maxillary Retrognathism. Indian J. Dent. Res. 2019, 30, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.J.; Netherway, D.J.; McGlaughlin, K.; David, D.J. Intracranial Volume Measurement of Sagittal Craniosynostosis. J. Clin. Neurosci. 2007, 14, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, J.B.; Gripp, K.W.; Stolle, C.A.; Steinberger, D.; Müller, U. Molecular Analysis of Patients with Synostotic Frontal Plagiocephaly (Unilateral Coronal Synostosis). Plast. Reconstr. Surg. 2004, 113, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.B.M.; Tang, G.H.; Xiong, H.; Hägg, U. PTHrP Regulates Chondrocyte Maturation in Condylar Cartilage. J. Dent. Res. 2003, 82, 627–631. [Google Scholar] [CrossRef]
- Baron, J.; Sävendahl, L.; De Luca, F.; Dauber, A.; Phillip, M.; Wit, J.M.; Nilsson, O. Short and Tall Stature: A New Paradigm Emerges. Nat. Rev. Endocrinol. 2015, 11, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Kamp, G.A.; Oostdijk, W. Towards a Rational and Efficient Diagnostic Approach in Children Referred for Growth Failure to the General Paediatrician. Horm. Res. Paediatr. 2019, 91, 223–240. [Google Scholar] [CrossRef]
- Naragond, A.; Kenganal, S.; Sagarkar, R.; Kumar, N.S.; Sugaradday. Diagnostic Limitations of Cephalometrics in Orthodontics-A Review. IOSR J. Dent. Med. Sci. 2012, 3, 30–35. [Google Scholar] [CrossRef]
- Davidopoulou, S.; Chatzigianni, A. Craniofacial Morphology and Dental Maturity in Children with Reduced Somatic Growth of Different Aetiology and the Effect of Growth Hormone Treatment. Prog. Orthod. 2017, 18, 10. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| GENETIC SYNDROMES ASSOCIATED WITH SHORT STATURE | DATA FROM LATERAL CEPHALOMETRIC RADIOGRAPHS | |||
|---|---|---|---|---|
| Skull | Anteroposterior Plane | Vertical Plane | Teeth | |
| Achondroplasia (1 in 15,000–40,000) | Skull deformities Pneumatized frontal sinuses Significantly reduced posterior cranial base length Acute cranial base angle Significantly reduced length of the cribriform plate of the ethmoidal bone Remarkably increased anterior sphenoidal length | Posteriorly placed and smaller maxilla and an anteriorly or normally placed mandible Skeletal class III that can result in an anterior cross-bite | Reduced upper anterior facial height Posterior tilt of the nasal floor (palatal plane) High coronoid process | Maxillary incisors labially proclined |
| Turner syndrome (1 in 2000–3000) | Reduced posterior cranial base length Increased cranial base angle Smaller and thiner calvarium Fused cervical vertebrae Inferiorly and anteriorly placed external acoustic meatus Smaller, more delicate and less pneumatized mastoid processes Large and excessively pneumatized sphenoidal sinuses Smaller sella turcica Premature calcification of the petroclinoid ligament Reduced facial/cerebral skull ratio | Reduced length of the maxilla Reduced length of the mandible Posteriorly positioned maxilla Posteriorly positioned mandible Posteriorly positioned chin Skeletal class II Transverse plane Facial asymmetry Posterior cross-bite as a result of the transversal dimension reduction of the maxilla | Hyperdivergent skeletal planes Reduced posterior facial height Increased anterior facial height | Occlusal plane angle is remarkably tilted Maxillary incisors lingually inclined Short teeth roots Remarkably low tongue position Pharyngeal airway space is narrower in all its dimensions |
| Noonan syndrome (1 in 1000–2500) | NA | Class I molar relationship and class III cuspid relationship, Class I skeletal relationship Class II skeletal relationship | Vertical mandibular growth pattern with hyperdivergent planes and increased vertical angles. Both an increased and a decreased vertical overjet have been reported | Labially inclined maxillary mandibular incisors Palatally inclined maxillary incisors and labially inclined mandibular incisors Lingually inclined mandibular incisors |
| Idiopathic growth hormone deficiency | Reduced anterior cranial base length | Reduced maxillary length and mandibular length | Reduced anterior facial height and ramus height | NA |
| Prader-Willi syndrome (1 in 10,000–20,000) | Reduced cranial base angle | Skeletal class II with posteriorly placed mandible and reduced mandibular and maxillary length Skeletal class III with anteriorly placed mandible | Vertical growth direction and increased anterior facial height In cases of skeletal class III, horizontal growth direction | Soft tissue excess In cases of skeletal class III, lingually inclined mandibular incisors and labially inclined maxillary incisors |
| Muenke syndrome (1 in 30,000) | Decreased intracranial volume Significantly reduced anterior cranial base and skull length Increased angle between cranial base and Frankfort horizontal plane Hypertelorism Frontal bossing | Reduced length of the maxilla and midface deficiency Reduced length of the mandible Posteriorly placed maxilla Transverse plane Increased facial width Significant skeletal asymmetry | Reduced upper and lower anterior facial height Hypedivergent skeletal planes Increased gonial angle Anterior open bite | NA |
| GENETIC SYNDROMES ASSOCIATED WITH SHORT STATURE | DATA FROM LATERAL CEPHALOMETRIC RADIOGRAPHS | |||
|---|---|---|---|---|
| Skull | Anteroposterior Plane | Vertical Plane | Teeth | |
| 22q11.2 deletion syndrome | Increased cranial base angle | Posteriorly placed mandible | NA | NA |
| 49, XXXXY syndrome (1 in 85,000–100,000) | NA | Anteriorly placed mandible | NA | Lingually inclined mandibular incisors |
| Catania brachydactylous type of acrofacial dysostosis | NA | No distinctive abnormalities | NA | NA |
| Beckwith–Wiedemann syndrome (1 in 11,000) | NA | Dental class I | Vertical growth pattern Anterior open bite | Macroglossia |
| Bloch–Sulzberger syndrome (1,2 in 100,000) | NA | Reduced maxillary length | Hyperdivergent skeletal planes | Lingually inclined maxillary incisors |
| Chronic acid sphingomyelinase deficiency (1 in 250,000) | NA | Posteriorly placed maxilla and mandible and skeletal class II | NA | Increased nasolabial angle Convex profile Retroinclination of maxillary and mandibular incisors |
| Cockayne syndrome (2–3 in a million) | Hypodevelopment | Posteriorly placed and shorter mandible and skeletal class II | NA | NA |
| Syndromic craniosynostosis with fused spheno-occipital synchondrosis (1 in 100,000) | Moderate and severe upward anterior cranial base inclination | Severe midface deficiency Higher percentage of severe Class III skeletal pattern | Severely hyperdivergent skeletal planes Severely forward condyle position | NA |
| Ectodermal dysplasia 1, hypohidrotic (1 in 10,000–100,000) | NA | Reduced length and posterior placement of the maxilla Anteriorly placed mandible with protruding chin Skeletal class III | Hyperdivergent skeletal planes Reduced anterior facial height Reduced upper anterior facial height | First maxillary molars located in higher positions |
| Ectodermal dysplasia anhidrotic or Rapp–Hodgkin syndrome | NA | Mildly to moderately reduced mandibular length with anterior mandibular placement Maxilla placed closer to the anterior cranial base | NA | NA |
| Ellis–Van Creveld syndrome (1 in 60,000–200,000) | NA | Skeletal class I or class II with posteriorly placed mandible Class III with anteriorly placed mandible, or posteriorly placed maxilla | Hyperdivergency of the skeletal planes, normal vertical growth direction, or even horizontal growth pattern | Mandibular and maxillary incisor retroclination Upper lip retrusion Lower lip retrusion Both concave and convex profiles have been reported |
| Hajdu–Cheney syndrome (less than 100 cases described) | Increased cranial base length Enlarged, elongated, and wide open sella turcica with slender clinoids | Posteriorly placed maxilla and mandible | NA | NA |
| Hallerman–Streiff syndrome (less than 200 people worldwide) | NA | Skeletal and dental class II due to shorter and posteriorly placed mandible | Vertical growth pattern with an opening of the gonial angle, a large anterior open bite, and an excessive increase in the lower anterior facial height | NA |
| Kabuki syndrome (1 in 32,000) | NA | Posteriorly placed maxilla and mandible with a skeletal Class I pattern | Increased lower anterior facial height and anterior open bite | NA |
| Klippel–Feil syndrome (1 in 40,000) | Fused cervical vertebrae | Skeletal class I | Vertical growth pattern | NA |
| Langer–Giedion syndrome (extremely rare) | NA | Posteriorly placed maxilla and mandible | NA | NA |
| Larsen syndrome (1 in 100,000) | Orbits positioned posteriorly relative to the anterior cranial base | Posteriorly positioned maxilla and mandible with skeletal Class III pattern Transverse plane Hypertelorism Narrow maxillary basal arch Reduced maxillary and mandibular dental arch widths | Increased vertical angles with a large Gonial angle Growth tendency of the mandible toward the posteroinferior direction | Mandibular primary incisors lingually inclined |
| Methylmalonic aciduria and homocystinuria (1 in 200,000) | Head rotated and bent towards the left shoulder, which is located in a lower position than the right one Horizontal planes of both maxillary bones converge towards the right | NA | NA | NA |
| Moebius syndrome (1 in 50,000–500,000) | NA | Posteriorly placed mandible with reduced length and skeletal class II | Increased maxillary height resulting in a vertical growth pattern | Proclined maxillary and mandibular incisors Protrusion of upper and lower lips Long upper lip |
| Congenital or childhood onset myotonic dystrophy type I (1 in 9000) | NA | Increased ANB angle and reduced facial angle | Hyperdivergent skeletal planes with mandibular plane angle and intermaxillary angle increased | NA |
| Rubinstein-Taybi syndrome (1 in 100,000 to 125,000) | Brachycephaly | Skeletal class II | NA | NA |
| Seckel syndrome (1 in 10,000) | Small skull with an extremely short anterior cranial base and maxillary length Differences in the morphology of the sella turcica observed between girls and boys | NA | NA | NA |
| Silver–Russell syndrome (1 in 30,000–100,000) | NA | Skeletal class II with posteriorly placed mandible Class I and III have also been reported | NA | NA |
| Simpson–Golabi–Behmel syndrome | Increased anterior cranial base length | Increased length of the maxilla and the mandible with a skeletal class III pattern | Increased lower anterior facial height | NA |
| Solitary Median Maxillary Central Incisor syndrome (1 in 50,000) | Hypoplastic sella turcica Cervical vertebral maturation (CVM) at stage CS2 | Skeletal class III with an anterior cross-bite as a result of reduced maxillary length and anteriorly placed mandible | Vertical growth pattern | Convex profile Airway patency Maxillary and mandibular incisal proclination |
| Treacher–Collins syndrome (1 in 50,000) | Reduced length of both the anterior and posterior cranial base and a reduced cranial base angle | Posteriorly placed maxilla with reduced length Posteriorly placed mandible with a characteristic reduction of the mandibular length Reduced maximum ramus width | Hyperdivergent skeletal planes and increased gonial angle Both the anterior and posterior facial heights are decreased | The maxillary and functional occlusal planes are tipped upwards posteriorly |
| Williams syndrome (1 in 7500–18,000) | Reduced anterior cranial base length | Posteriorly placed chin | Hypedivergent skeletal planes Unusual proportion of upper to lower anterior facial height and posterior to anterior facial height | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paltoglou, G.; Ziakas, N.; Chrousos, G.P.; Yapijakis, C. Cephalometric Evaluation of Children with Short Stature of Genetic Etiology: A Review. Children 2024, 11, 792. https://doi.org/10.3390/children11070792
Paltoglou G, Ziakas N, Chrousos GP, Yapijakis C. Cephalometric Evaluation of Children with Short Stature of Genetic Etiology: A Review. Children. 2024; 11(7):792. https://doi.org/10.3390/children11070792
Chicago/Turabian StylePaltoglou, George, Nickolas Ziakas, George P. Chrousos, and Christos Yapijakis. 2024. "Cephalometric Evaluation of Children with Short Stature of Genetic Etiology: A Review" Children 11, no. 7: 792. https://doi.org/10.3390/children11070792
APA StylePaltoglou, G., Ziakas, N., Chrousos, G. P., & Yapijakis, C. (2024). Cephalometric Evaluation of Children with Short Stature of Genetic Etiology: A Review. Children, 11(7), 792. https://doi.org/10.3390/children11070792