

Heterogenic Genetic Background of Distal Arthrogryposis—Review of the Literature and Case Report
 , ,
, ,
Abstract
:1. Introduction
1.1. Epidemiology, Characteristics, and Genetics
- Amyoplasia: The symmetric absence of muscle development, mostly in the limbs. The etiology is unclear. The upper limb is rotated at birth with an extended elbow and a flexed wrist. Clubfoot of varying severity is seen. Associated anomalies include frontal hemangioma and genital and finger hypoplasia. Gastroschisis is present in 10% of cases. Affected children have normal intelligence and no craniofacial abnormalities.
- Distal-type arthrogryposis (DA): An autosomal dominant inherited disorder affecting mainly the lower limbs, excluding the large joints. No clear neuro- or myogenic abnormalities are underlying. The upper limb is affected to a lesser extent, with ulnar deviation in the form of camptodactyly. The phenotype is highly heterogeneous.
- Systemic connective tissue diseases: Contractural arachnodactyly, Beals syndrome, Larsen syndrome, and Ehlers–Danlos syndrome. Ligamentous disorders lead to restricted joint motion.
- c/1 Beals syndrome: The inheritance of this syndrome is autosomal dominant, characterized by contractures, camptodactyly, arachnodactyly, scoliosis, and hypoplasia. The most commonly affected gene is FBN2 [7].
- c/2 Larsen syndrome: The inheritance of this syndrome is autosomal dominant or sporadic, characterized by joint dislocation, spine anomalies, and characteristic facial features. The most commonly affected gene is FLNB [8].
- c/3 Ehlers–Danlos syndrome: This is a heterogenous disease with 14 subtypes. Common features include joint hypermobility, skin abnormalities, and wound-healing problems. Arthrogryposis can happen but is not present in all cases [9].
- d.
- “Multiple pterygium” syndrome: There are lethal and non-lethal (Escobar-type) forms of this syndrome. Its genetics and phenotypic presentation are highly heterogeneous.
- Lethal form: This form involves IUGR/SGR (intrauterine growth retardation/small for gestational age), contractures, pterygia, and a dysmorphic face. Affected individuals are stillborn or do not survive the perinatal period. Camptodactyly may occur with or without syndactyly. Minor facial anomalies, ptosis, and antimongoloid eyes are often seen.
- Non-lethal/Escobar type: This form involves scoliosis, cryptorchidism, and facial involvement with normal intelligence
- e.
- Oligohydramnios/external factors
1.2. Ultrasound Findings
1.3. Antenatal Care
1.4. Prognosis and Treatment
1.5. Distal Arthrogryposis (DA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Clinical Features | Gene | Inheritance |
|---|---|---|---|
| Distal arthrogryposis type 1 | Camptodactyly and clubfoot | TPM2 | AD |
| Distal arthrogryposis type 2A (Freeman–Sheldon syndrome) | Contractures of fingers and toes, kyphosis, scoliosis, and whistling face | MYH3 | AD |
| Distal arthrogryposis type 2B (Sheldon-Hall syndrome) | Features similar to distal type 1 and type 2A with distal joint contractures in the limbs, triangular face, downward-slanting palpebral fissures, small mouth, and high-arched palate | TNNI2, TNNT3, MYH3, TMP2 | AD |
| Distal arthrogryposis type 3 (Gordone syndrome) | Short stature, cleft palate, and palatoschisis | PIEZO2 | AD |
| Distal arthrogryposis type 4 | Contractures and severe scoliosis | N/A | AD |
| Distal arthrogryposis type 5 | Limitation of eye movement (opthalmoplegia), ptosis and strabismus | PIEZO2 | AD |
| Distal arthrogryposis type 6 | Sensorineural hearing loss | N/A | N/A |
| Distal arthrogryposis type 7 | Trismus-pseudocamptodactyly, short stature, and shortened paralyzed muscles | MYH8 | AD |
| Distal arthrogryposis type 8 | Autosomal dominant multiple pterygium syndrome | MYH3 | AD |
| Distal arthrogryposis type 9 | Congenital contractural arachnodactyly, phenotypes similar to those of Marfan syndrome, but without cardiovascular abnormalities | FBN2 | AD |
| Distal arthrogryposis type 10 | Congenital plantar contractures | 2q | AD |
| Distal arthrogryposis type 11 | Camptodactyly, absent flexion creases, and limited forearm supination | MET | AD |
| Distal arthrogryposis type 12 | Congenital contractures (small joints of the fingers and toes), contractures of the knees and Achilles tendons, spinal stiffness, scoliosis, and orthodontic abnormalities | ADAMTS15 | AR |
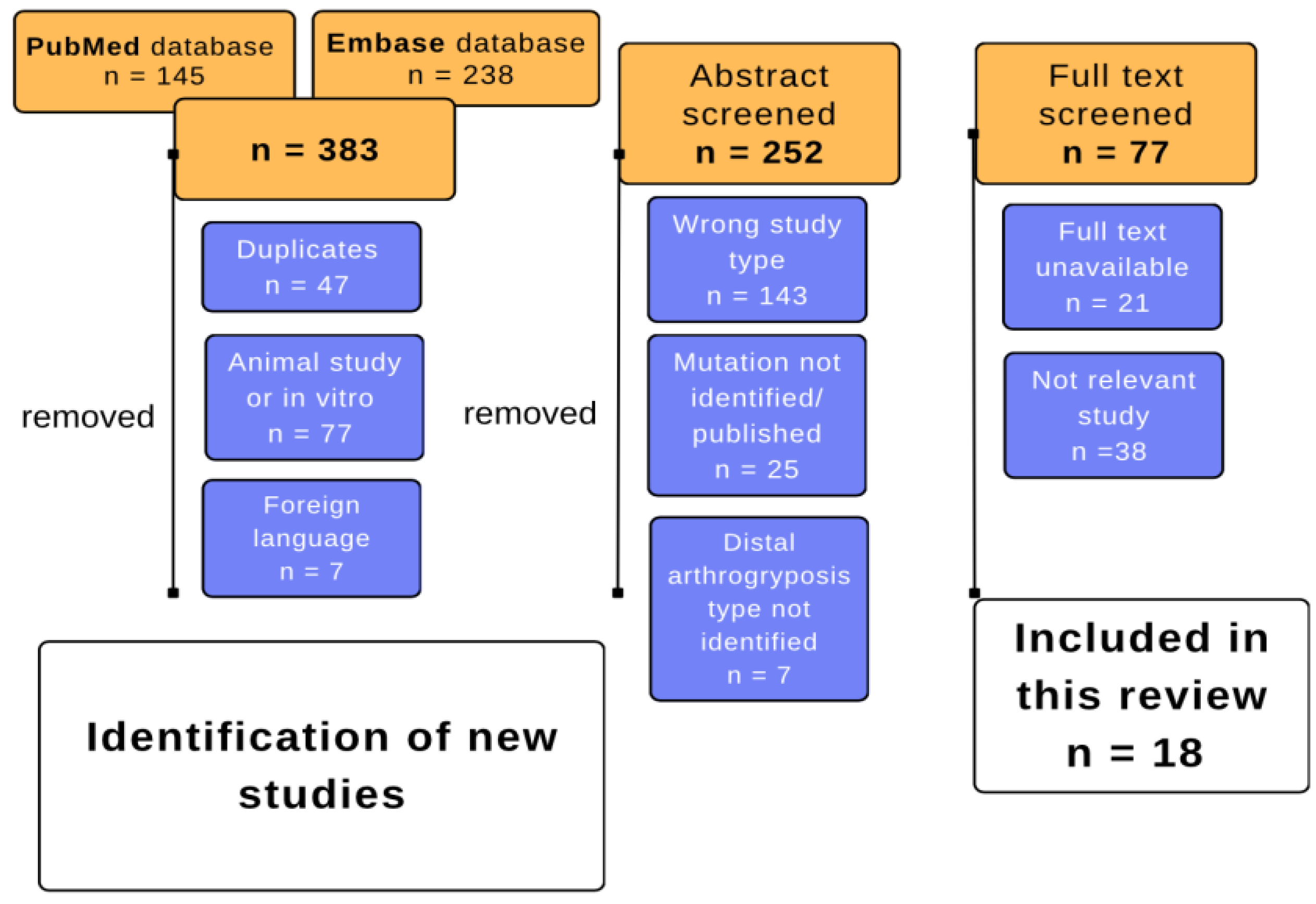
2. Materials and Methods
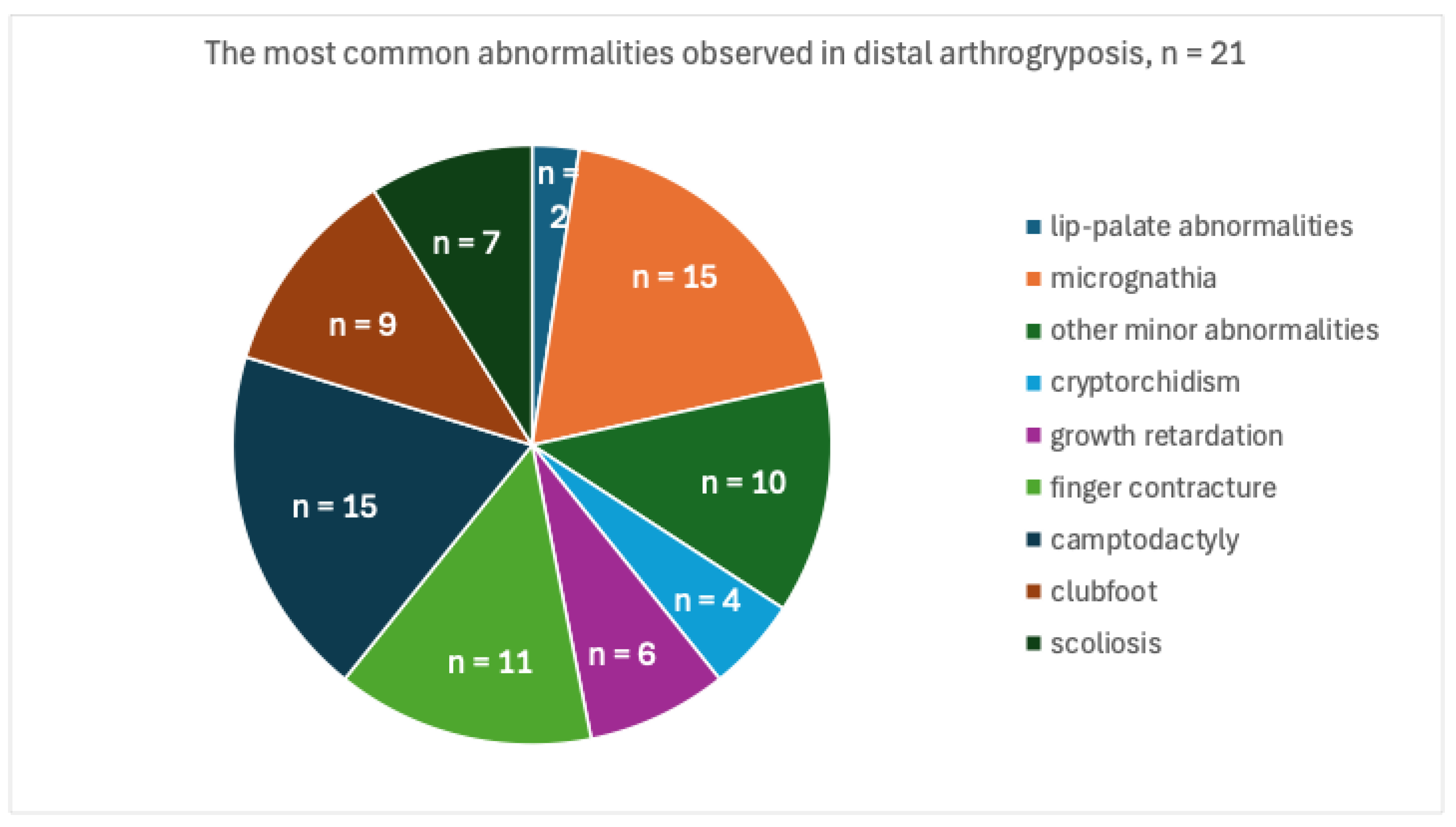
3. Results
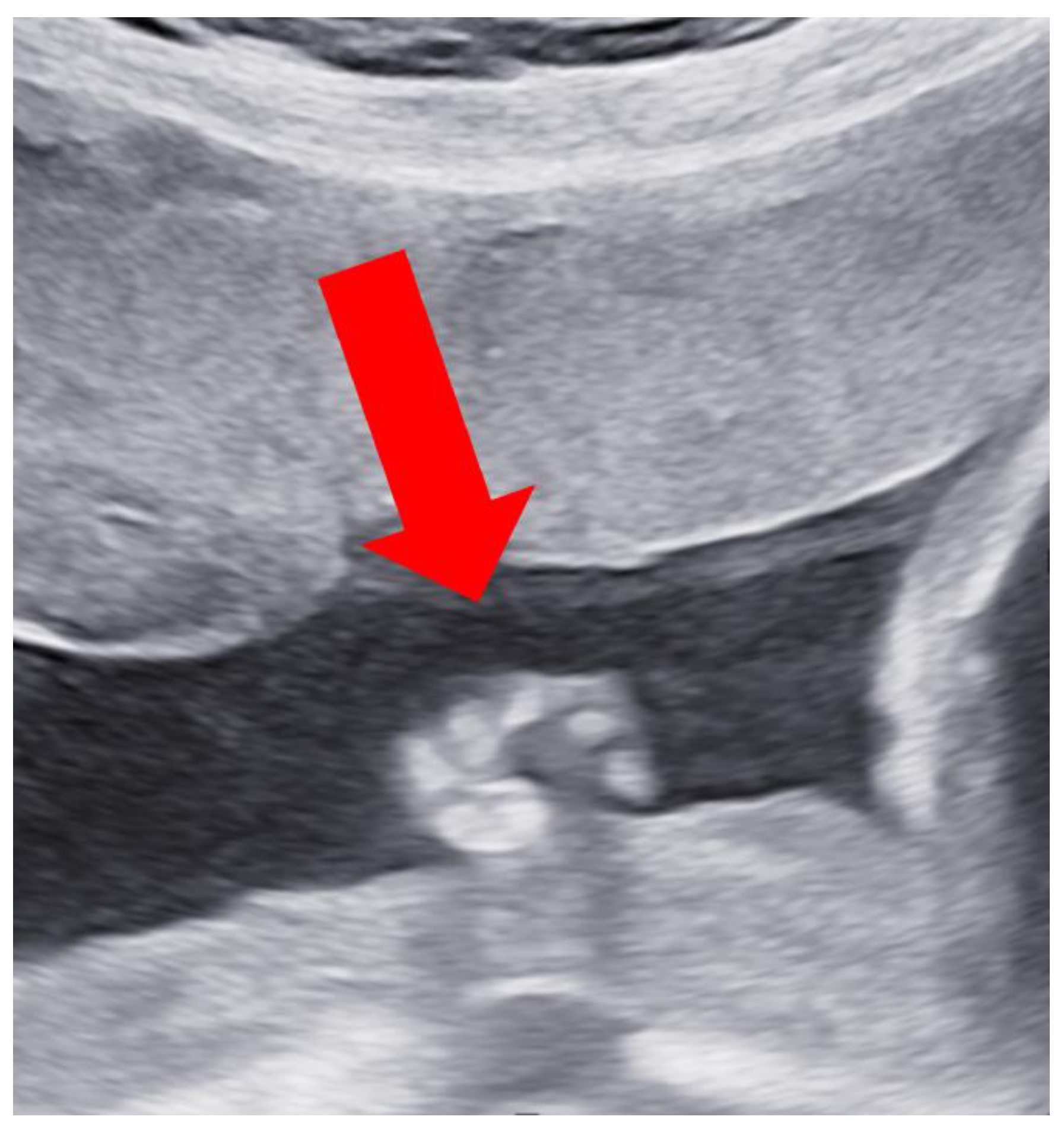
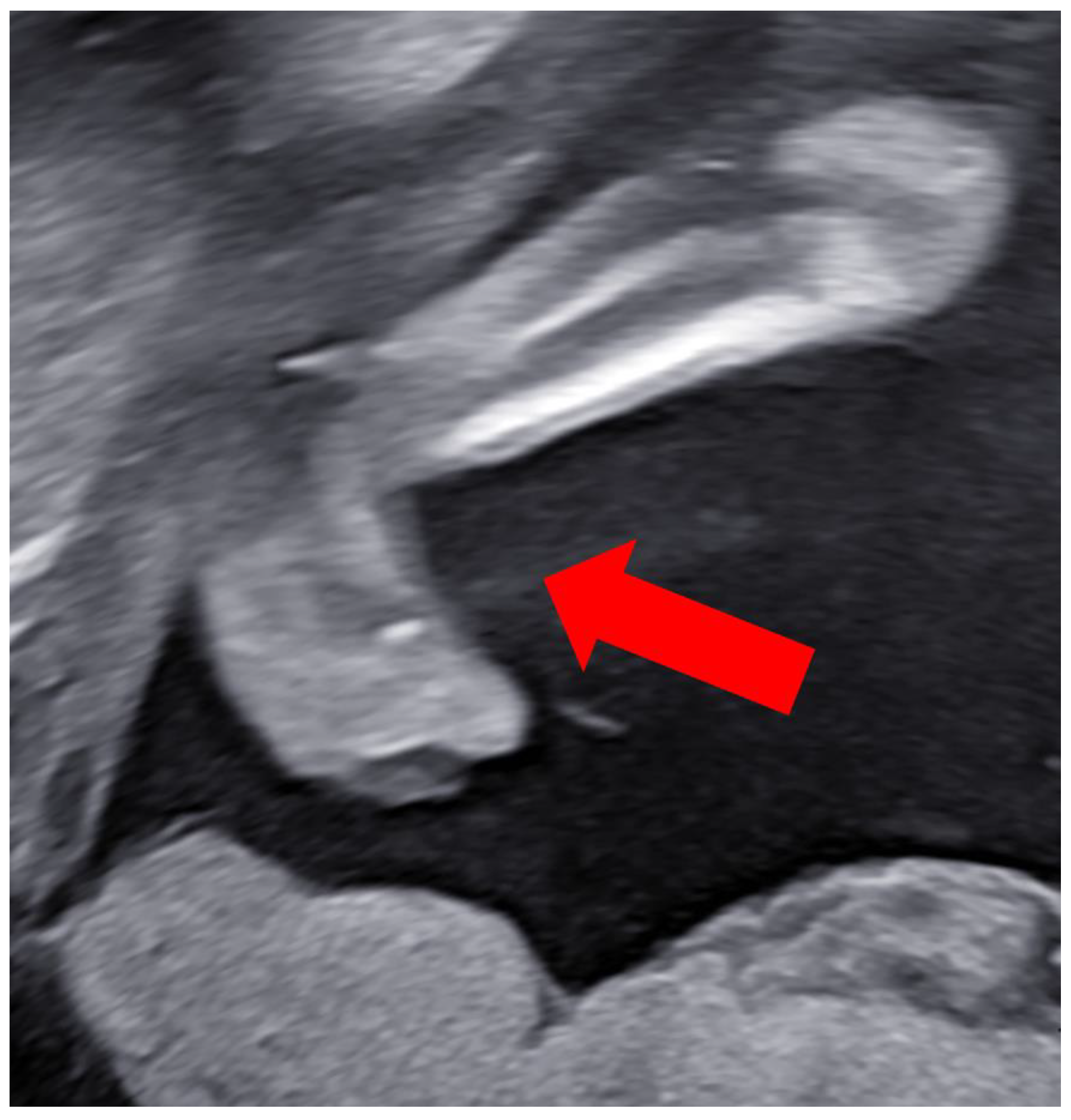
Case Report
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rac, M.W.F.; McKinney, J.; Gandhi, M. Arthrogryposis. Am. J. Obstet. Gynecol. 2019, 221, B7–B9. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014, 57, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.; Johnson, A.; Dobbs, M.B.; Gurnett, C.A. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes 2021, 12, 943. [Google Scholar] [CrossRef]
- Kowalczyk, B.; Feluś; J. Arthrogryposis: An update on clinical aspects, etiology, and treatment strategies. Arch. Med. Sci. 2016, 12, 10–24. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M. Neurogenic arthrogryposis and the power of phenotyping. Neuromuscul. Disord. 2021, 31, 1062–1069. [Google Scholar] [CrossRef]
- Skaria, P.; Dahl, A.; Ahmed, A. Arthrogryposis multiplex congenita in utero: Radiologic and pathologic findings. J. Matern. Fetal Neonatal Med. 2019, 32, 502–511. [Google Scholar] [CrossRef]
- Tunçbilek, E.; Alanay, Y. Congenital contractural arachnodactyly (Beals syndrome). Orphanet. J. Rare Dis. 2006, 1, 20. [Google Scholar] [CrossRef]
- Siafaka, A.; Angelis, S.; Piagkou, M.; Apostolopoulos, A.; Troupis, T.; Filippou, D. Larsen Syndrome and Associated Spinal Deformities. Cureus 2023, 15, e41655. [Google Scholar] [CrossRef]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Wahlig, B.; Poppino, K.; Jo, C.H.; Rathjen, K. Arthrogryposis multiplex congenita: A 28-year retrospective study. Dev. Med. Child. Neurol. 2022, 64, 476–480. [Google Scholar] [CrossRef]
- Laquerriere, A.; Jaber, D.; Abiusi, E.; Maluenda, J.; Mejlachowicz, D.; Vivanti, A.; Dieterich, K.; Stoeva, R.; Quevarec, L.; Nolent, F.; et al. Phenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita. J. Med. Genet. 2022, 59, 559–567. [Google Scholar] [CrossRef]
- Kiefer, J.; Hall, J.G. Gene ontology analysis of arthrogryposis (multiple congenital contractures). Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 310–326. [Google Scholar] [CrossRef]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy: Outcomes and Lessons Learned. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Barlow, D.P.; Gautel, M.; Gibson, T.; Holt, J.; Hsieh, C.L.; Francke, U.; Leonard, K.; Wardale, J.; Whiting, A.; et al. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature 1990, 1990. 345, 273–276. [Google Scholar] [CrossRef]
- Eu, J.P.; Sun, J.; Xu, L.; Stamler, J.S.; Meissner, G. The skeletal muscle calcium release channel: Coupled O2 sensor and NO signaling functions. Cell 2000, 102, 499–509. [Google Scholar] [CrossRef]
- Ahangari, N.; Gholampour-Faroji, N.; Doosti, M.; Ghayour Mobarhan, M.; Shahrokhzadeh, S.; Karimiani, E.G.; Hasani-Sabzevar, B.; Torbati, P.N.; Haddad-Mashadrizeh, A. ECEL1 novel mutation in arthrogryposis type 5D: A molecular dynamic simulation study. Mol. Genet. Genomic Med. 2023, 11, e2153. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Barth, P.G.; Van Lookeren Campagne, W.; De Visser, M.; Dingemans, K.P. A family with severe X-linked arthrogryposis. Eur. J. Pediatr. 1991, 150, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Stedman, H.; Browning, K.; Oliver, N.; Oronzi-Scott, M.; Fischbeck, K.; Sarkar, S.; Sylvester, J.; Schmickel, R.; Wang, K. Nebulin cDNAs detect a 25-kilobase transcript in skeletal muscle and localize to human chromosome 2. Genomics 1988, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Teuling, E.; van Dis, V.; Wulf, P.S.; Haasdijk, E.D.; Akhmanova, A.; Hoogenraad, C.C.; Jaarsma, D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 2008, 17, 2849–2862. [Google Scholar] [CrossRef]
- Bhat, M.A.; Rios, J.C.; Lu, Y.; Garcia-Fresco, G.P.; Ching, W.; St Martin, M.; Li, J.; Einheber, S.; Chesler, M.; Rosenbluth, J.; et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 2001, 30, 369–383. [Google Scholar] [CrossRef]
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C.S. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007, 21, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Jones, W.D.; McIntyre, R.; Sanchez-Andrade, G.; Sanderson, M.; Stephenson, J.D.; Jones, C.P.; Handsaker, J.; Gallone, G.; Bruntraeger, M.; et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science 2018, 362, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yu, X. Arthrogryposis multiplex congenita: Classification, diagnosis, perioperative care, and anesthesia. Front. Med. 2017, 11, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Cachecho, S.; Boruff, J.; Wong, T.; Lacombe, F.; Dahan-Oliel, N. Psychosocial wellbeing among children and adults with arthrogryposis: A scoping review. Health Qual. Life Outcomes 2021, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Stiene, D.; Song, T.; Sadayappan, S. Distal Arthrogryposis and Lethal Congenital Contracture Syndrome—An Overview. Front. Physiol. 2020, 11, 689. [Google Scholar] [CrossRef]
- Pehlivan, D.; Bayram, Y.; Gunes, N.; Coban Akdemir, Z.; Shukla, A.; Bierhals, T.; Tabakci, B.; Sahin, Y.; Gezdirici, A.; Fatih, J.M.; et al. The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am. J. Hum. Genet. 2019, 105, 132–150. [Google Scholar] [CrossRef]
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A review and update. J. Bone Joint Surg. Am. 2009, 9 (Suppl. 4), 40–46. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/ (accessed on 8 May 2024).
- Bamshad, M.; Bohnsack, J.F.; Jorde, L.B.; Carey, J.C. Distal arthrogryposis type 1: Clinical analysis of a large kindred. Am. J. Med. Genet. 1996, 65, 282–285. [Google Scholar] [CrossRef]
- Gurnett, C.A.; Desruisseau, D.M.; McCall, K.; Choi, R.; Meyer, Z.I.; Talerico, M.; Miller, S.E.; Ju, J.S.; Pestronk, A.; Connolly, A.M.; et al. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010, 19, 1165–1173. [Google Scholar] [CrossRef]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Kezele, P.R.; Shively, K.M.; Carey, J.C.; Regnier, M.; Bamshad, M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. A 2013, 161a, 550–555. [Google Scholar] [CrossRef]
- Toydemir, R.M.; Bamshad, M.J. Sheldon-Hall syndrome. Orphanet J. Rare Dis. 2009, 4, 11. [Google Scholar] [CrossRef]
- Dieterich, K.; Quijano-Roy, S.; Monnier, N.; Zhou, J.; Fauré, J.; Smirnow, D.A.; Carlier, R.; Laroche, C.; Marcorelles, P.; Mercier, S.; et al. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum. Mol. Genet. 2013, 22, 1483–1492. [Google Scholar] [CrossRef]
- Chong, J.X.; Childers, M.C.; Marvin, C.T.; Marcello, A.J.; Gonorazky, H.; Hazrati, L.N.; Dowling, J.J.; Amrani, F.A.; Alanay, Y.; Nieto, Y.; et al. Variants in ACTC1 underlie distal arthrogryposis accompanied by congenital heart defects. Hum. Genet. Genom. Adv. 2023, S4, 100213. [Google Scholar] [CrossRef]
- Zapata-Aldana, E.; Al-Mobarak, S.B.; Karp, N.; Campbell, C. Distal arthrogryposis type 5 and PIEZO2 novel variant in a Canadian family. Am. J. Med. Genet. A 2019, 179, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Sloma, R.; Pizem, H.; Fedida, A.; Kalfon, L.; Ovadia, R.; Segal, Z.; Kassif, Y.; Falik Zaccai, T. Long term ophthalmic complications of distal arthrogryposis type 5D. Ophthalmic Genet. 2023, 44, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Gowda, M.; Mohan, S.; Ramesh, D.; Chinta, N. Distal arthrogryposis type 5D in a South Indian family caused by novel deletion in ECEL1 gene. Clin. Dysmorphol. 2021, 30, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Karaca, E.; Coban Akdemir, Z.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S.; et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef]
- Pollazzon, M.; Caraffi, S.G.; Faccioli, S.; Rosato, S.; Fodstad, H.; Campos-Xavier, B.; Soncini, E.; Comitini, G.; Frattini, D.; Grimaldi, T.; et al. Clinical and Genetic Findings in a Series of Eight Families with Arthrogryposis. Genes 2021, 13, 29. [Google Scholar] [CrossRef]
- Wang, W.B.; Kong, L.C.; Zuo, R.T.; Kang, Q.L. Identification of a novel pathogenic mutation of the MYH3 gene in a family with distal arthrogryposis type 2B. Mol. Med. Rep. 2020, 21, 438–444. [Google Scholar] [CrossRef]
- Dabaj, I.; Carlier, R.Y.; Dieterich, K.; Desguerre, I.; Faure, J.; Romero, N.B.; Trang, W.; Quijano-Roy, S.; Germain, D.P. Diagnostic work-up and phenotypic characteristics of a family with variable severity of distal arthrogryposis type 2B (Sheldon-Hall syndrome) and TNNT3 pathogenic variant. Front. Genet. 2022, 13, 955041. [Google Scholar] [CrossRef] [PubMed]
- Serra, G.; Antona, V.; Cannata, C.; Giuffrè, M.; Piro, E.; Schierz, I.A.M.; Corsello, G. Distal Arthrogryposis type 5 in an Italian family due to an autosomal dominant gain-of-function mutation of the PIEZO2 gene. Ital. J. Pediatr. 2022, 48, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, W.Q.; Wang, S.W.; Wang, S.X.; Yu, M.; Guo, Q.; Yu, Y.D. Identification of a novel pathogenic variant in the MYH3 gene in a five-generation family with CPSFS1A (Contractures, Pterygia, and Spondylocarpotarsal Fusion Syndrome 1A). Mol. Genet. Genom. Med. 2020, 8, e1440. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nong, T.; Li, Y.; Li, X.; Li, Z.; Lv, H.; Xu, H.; Li, J.; Zhu, M. A TNNI2 variant c.525G>T causes distal arthrogryposis in a Chinese family. Mol. Genet. Genomic Med. 2022, 10, e2042. [Google Scholar] [CrossRef] [PubMed]
- Alesi, V.; Sessini, F.; Genovese, S.; Calvieri, G.; Sallicandro, E.; Ciocca, L.; Mingoia, M.; Novelli, A.; Moi, P. A New Intronic Variant in ECEL1 in Two Patients with Distal Arthrogryposis Type 5D. Int. J. Mol. Sci. 2021, 22, 2106. [Google Scholar] [CrossRef] [PubMed]
- Sandaradura, S.A.; Bournazos, A.; Mallawaarachchi, A.; Cummings, B.B.; Waddell, L.B.; Jones, K.J.; Troedson, C.; Sudarsanam, A.; Nash, B.M.; Peters, G.B.; et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum. Mutat. 2018, 39, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, B.; Han, W.; Zhao, N.; Liu, W.; Sui, Y.; Wang, Y.; Lu, Y.; Wang, H.; Li, J.; et al. Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS ONE 2015, 10, e0117158. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Steichen-Gersdorf, E.; Krabichler, B.; Petersen, B.S.; Weber, U.; Schmidt, W.M.; Zschocke, J.; Müller, T.; Bittner, R.E.; Janecke, A.R. Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: A genotype-phenotype correlation. Eur. J. Hum. Genet. 2017, 25, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Mbwasi, R.M.; Kinabo, G.; Kamsteeg, E.J.; Hamel, B.C.; Dekker, M.C.J. Freeman-Sheldon Syndrome: First Molecularly Confirmed Case from Sub-Saharan Africa. Case Rep. Genet. 2017, 2017, 9327169. [Google Scholar] [CrossRef]
- Iyer, A.; Lauerova, B.; Mariano, J.; Haberlová, J.; Lassuthova, P.; Zidkova, J.; Wright, N.T.; Kontrogianni-Konstantopoulos, A. Compound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations. Gene 2024, 910, 148339. [Google Scholar] [CrossRef]
- Jing, S.; Peng, M.; He, Y.; Hua, Y.; Li, J.; Li, Y. A novel compound heterozygous variant of ECEL1 induced joint dysfunction and cartilage degradation: A case report and literature review. Front. Neurol. 2024, 15, 1343025. [Google Scholar] [CrossRef]
- Neissi, M.; Sheikh-Hosseini, M.; Mohammadi-Asl, J.; Al-Badran, A.I. A novel heterozygous TPM2 gene mutation (c.456G>C; p.Lys152Asn) in an Iranian family affected by distal arthrogryposis type 1: A case report. Egypt. J. Med. Hum. Genet. 2022, 23, 49. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Sung, K.H.; Lee, S.H.; Kim, N.; Cho, T.J. Orthopaedic manifestations and treatment outcome of two siblings with Escobar syndrome and homozygous mutations in the CHRNG gene. J. Pediatr. Orthop. B 2015, 24, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; Alaee, F.; Desruisseau, D.; Boehm, S.; Dobbs, M.B. Skeletal muscle contractile gene (TNNT3, MYH3, TPM2) mutations not found in vertical talus or clubfoot. Clin. Orthop. Relat. Res. 2009, 467, 1195–1200. [Google Scholar] [CrossRef]
- Laquérriere, A.; Maluenda, J.; Camus, A.; Fontenas, L.; Dieterich, K.; Nolent, F.; Zhou, J.; Monnier, N.; Latour, P.; Gentil, D.; et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum. Mol. Genet. 2014, 23, 2279–2289. [Google Scholar] [CrossRef]
- Vora, N.L.; Gilmore, K.; Brandt, A.; Gustafson, C.; Strande, N.; Ramkissoon, L.; Hardisty, E.; Foreman, A.K.M.; Wilhelmsen, K.; Owen, P.; et al. An approach to integrating exome sequencing for fetal structural anomalies into clinical practice. Genet. Med. 2020, 22, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Daly, S.B.; Shah, H.; O’Sullivan, J.; Anderson, B.; Bhaskar, S.; Williams, S.; Al-Sheqaih, N.; Mueed Bidchol, A.; Banka, S.; Newman, W.G.; et al. Exome Sequencing Identifies a Dominant TNNT3 Mutation in a Large Family with Distal Arthrogryposis. Mol. Syndromol. 2014, 5, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Griffet, J.; Dieterich, K.; Bourg, V.; Bourgeois, E. Amyoplasia and distal arthrogryposis. Orthop. Traumatol. Surg. Res. 2021, 107, 102781. [Google Scholar] [CrossRef]
- Sung, S.S.; Brassington, A.M.; Krakowiak, P.A.; Carey, J.C.; Jorde, L.B.; Bamshad, M. Mutations in TNNT3 cause multiple congenital contractures: A second locus for distal arthrogryposis type 2B. Am. J. Hum. Genet. 2003, 73, 212–214. [Google Scholar] [CrossRef]
- Mao, C.; Baumgartner, A.P.; Jha, P.K.; Huang, T.H. Sarkar, S. Assignment of the human fast skeletal troponin T gene (TNNT3) to chromosome 11p15.5: Evidence for the presence of 11pter in a monochromosome 9 somatic cell hybrid in NIGMS mapping panel 2. Genomics 1996, 31, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ochala, J. Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. J. Mol. Med. 2008, 86, 1197–1204. [Google Scholar] [CrossRef] [PubMed]

| Gene Name | Protein Encoded | Locus | Mechanism of Action | Source |
|---|---|---|---|---|
| TTN | TITIN | 2q31.2 | Titin helps to form a connection between the thick filament and the Z lineage in the sarcomere. It is also required for the interaction of several sarcomeric proteins. | [14] |
| CHRNG | ACETYLCHOLINE RECEPTOR | 2q37.1 | Acetylcholine receptor-dependent mechanisms. | [11] |
| RYR1 | RYANODINE RECEPTOR 1 | 19q13.2 | Cellular calcium homeostasis, redox system. | [15] |
| ECEL1 | ENDOTHELIN-CONVERTING ENZYME-LIKE 1 | 2q37.1 | Membrane-bound metalloprotease and endoprotease, involved in neuropeptide- and peptide-regulated hormone activity. Found in the spine, and lungs, but most highly expressed in the central nervous system. | [16] |
| ZC4H2 | ZINC FINGER C4H2 DOMAIN-CONTAINING PROTEIN | Xq11.2 | In a mouse model, the protein is expressed mainly in the hippocampal and dentate processes in the postsynaptic terminal of excitatory neurons. | [17] |
| NEB | NEBULIN | 2q23.3 | Nebulin is a large protein of the cytoskeletal matrix. In vertebrae, nebulin is responsible for 3–4% of myofibrillar proteins. | [18] |
| BICD2 | BICD CARGO ADAPTOR 2 | 9q22.31 | Involved in the function of dynein/dynactin in motoneurons. | [19] |
| CNTNAP1 | CONTACTIN-ASSOCIATED PROTEIN 1 | 17q21.2 | Part of the paranodal junction required for myelination of rapidly conducting neurons. | [20] |
| MAGEL2 | MAGE-LIKE 2 | 15q11.2 | Ubiquitin ligase required for endosomal protein recycling. | [21] |
| TPM2 | TROPOMYOSIN 2 | 9p13.3 | Switching the location of the actin–tropomyosin interface between active and relaxed states. | [22] |
| Article | Sex | CMA | Prenatally Diagnosed | Gene | Mutation | Mutation Effect | Locus | |
|---|---|---|---|---|---|---|---|---|
| Zapata-Aldana et al., 2019 [36] | N/A | DA5D | no | PIEZO2 | heterozygous | c.8068A>C (p.Ser2690Arg) | LOF | 18p11.22-p11.21 |
| Cohen et al., 2023 [37] | female | DA5D | no | ECEL1 | homozygous | c.110_155del p.(phe37cysfs * 151) | deletion | 2q37.1 |
| Gowda et al., 2021 [38] | male | DA5D | yes | ECEL1 | homozygous | c.535A>G (p. Lys179Glu) | missense | 2q37.1 |
| Bayram et al., 2016 [27,39] | N/A | DA5D | N/A | ECEL1 | homozygous | c.1147C>T; p.Gln383X | missense | 2q37.1 |
| N/A | DA5D | N/A | ECEL1 | homozygous | c.2023G>A; p.Ala675Thr | missense | 2q37.1 | |
| N/A | DA5D | N/A | ECEL1 | homozygous | c.1210C>T; p.Arg404Cys | missense | 2q37.1 | |
| Pollazzon et al., 2021 [40] | male | DA1 | yes | TPM2 | heterozygous | c.463G>A, p.(A155T) | missense | 9p13.3 |
| female | DA1 | no | TPM2 | heterozygous | c.463G>A, p.(A155T) | missense | 9p13.3 | |
| female | DA1 | N/A | TPM2 | heterozygous | c.463G>A, p.(A155T) | missense | 9p13.3 | |
| male | DA1 | yes | TNNT3 | heterozygous | c.187C>T, p.(R63C) | missense | 11p15.5 | |
| female | DA1 | yes | TNNT3 | heterozygous | c.187C>T, p.(R63C) | missense | 11p15.5 | |
| female | DA1 | yes | TNNT3 | heterozygous | c.187C>T, p.(R63C) | missense | 11p15.5 | |
| female | DA2B | yes | TNNI2 | heterozygous | c.499_501del, p.(E167del) | deletion | 11p15.5 | |
| female | DA2B | no | TNNI2 | heterozygous | c.499_501del, p.(E167del) | deletion | 11p15.5 | |
| male | DA5D | no | ECEL1 | heterozygous | c.[1630C>T];[1700C>G], p.[(R544C)]; | 11p15.5 | ||
| Wang et al., 2020 [41] | male | DA2B | no | MYH3 | heterozygous | c.2506A>G (p.K836E) | missense | 17p13.1 |
| female | DA2B | no | MYH3 | heterozygous | c.2506A>G (p.K836E) | missense | 17p13.1 | |
| male | DA2B | no | MYH3 | heterozygous | c.2506A>G (p.K836E) | missense | 17p13.1 | |
| Dabaj, Carlier et al. 2022) [42] | male | DA2B | N/A | TNNT3 | heterozygous | c.187C>T | missense | 11p15.5 |
| female | DA2B | no | TNNT3 | heterozygous | c.187C>T | missense | 11p15.5 | |
| female | DA2B | no | TNNT3 | heterozygous | c.187C>T | missense | 11p15.5 | |
| male | DA2B | no | TNNI2 | heterozygous | c.525G>T: p.K175N | missense | 11p15.5 | |
| Serra et al., 2022 [43] | male | DA5D | second trimester | PIEZO2 | heterozygous | c.8181_8183delAGA | GOF | 18p11.22-p11.21 |
| Zhang et al., 2020 [44] | male | DA5D | no | ECEL1 | homozygous | c.69C>A, p.C23 | missense | 2q37.1 |
| Li et al., 2022 [45] | male | DA5D | no | ECEL1 | homozygous | c.1507-9G>A | missense | 2q37.1 |
| Alesi et al., 2021 [46] | female | DA5D | decreased fetal movement | ECEL1 | homozygous | c.1507-9G>A | missense | 2q37.1 |
| female | DA5D | decreased fetal movement | ECEL1 | homozygous | c.1507-9G>A | missense | 2q37.1 | |
| Sandaradura et al., 2018 [47] | male | NM-DA | yes | TNNT3 | homozygous | c.681+1G>A | missense | 11p15.5 |
| Li et al., 2015 [48] | N/A | DA2 | N/A | MYBPC1 | N/A | c.1075G>A | missense | 12q23.2 |
| N/A | DA2 | N/A | MYBPC1 | N/A | c.956C>T | missense | 12q23.2 | |
| Baumann et al., 2017 [49] | male | NM-DA | decreased fetal movement | SYNE1 | homozygous | c.26236C>T | premature stop | 6q25.2 |
| Ali et al., 2017 [50] | male | DA2B | no | MYH3 | heterozygous | c.2015G>A | missense | 17p13.1 |
| Iyer et al., 2024 [51] | N/A | DA1 | yes | MYBPC1 | heterozygous | c.2486_2492del | deletion | 12q23.2 |
| Jung et al., 2024 [52] | male | DA5D | yes | ECEl1 | heterozygous | c.110_155del | deletion (frameshift) | 2q37.1 |
| Neissi et al., 2022 [53] | male | DA1 | N/A | TPM2 | N/A | c.456G>C | missense | 9p13.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illés, A.; Pikó, H.; Bartek, V.; Szepesi, O.; Rudas, G.; Benkő, Z.; Harmath, Á.; Kósa, J.P.; Beke, A. Heterogenic Genetic Background of Distal Arthrogryposis—Review of the Literature and Case Report. Children 2024, 11, 861. https://doi.org/10.3390/children11070861
Illés A, Pikó H, Bartek V, Szepesi O, Rudas G, Benkő Z, Harmath Á, Kósa JP, Beke A. Heterogenic Genetic Background of Distal Arthrogryposis—Review of the Literature and Case Report. Children. 2024; 11(7):861. https://doi.org/10.3390/children11070861
Chicago/Turabian StyleIllés, Anett, Henriett Pikó, Virág Bartek, Olívia Szepesi, Gábor Rudas, Zsófia Benkő, Ágnes Harmath, János Pál Kósa, and Artúr Beke. 2024. "Heterogenic Genetic Background of Distal Arthrogryposis—Review of the Literature and Case Report" Children 11, no. 7: 861. https://doi.org/10.3390/children11070861