
Confirming the Suitability of a Gentamicin Dosing Strategy in Neonates Using the Population Pharmacokinetic Approach with Truncated Sampling Duration
 , ,
, ,
Abstract
:
1. Introduction
2. Methods
2.1. Study Design and Setting
2.2. Participants
2.3. Drug Administration and Pharmacokinetic Sampling
2.4. Data Analysis
2.4.1. General Procedure
2.4.2. Analysis Software
2.4.3. Structural Model Identification
2.4.4. Covariate Analysis
2.4.5. Shrinkage and Reliability of Parameter Estimates
2.5. Predictive Performance
3. Results
3.1. Patient Characteristics
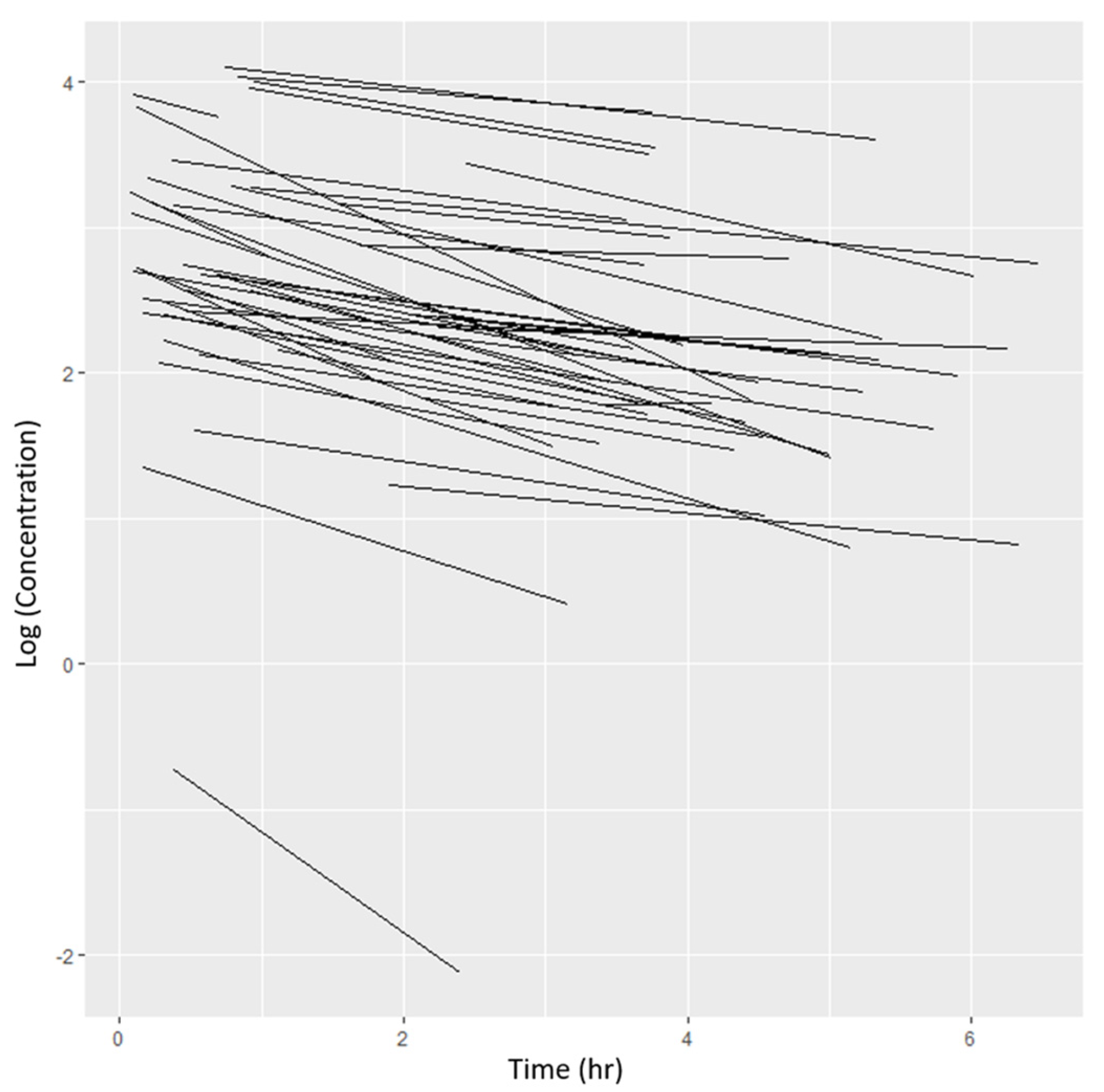
3.2. Pharmacokinetic Sampling
3.3. Base Model Identification
3.4. Population Pharmacokinetic Model Development
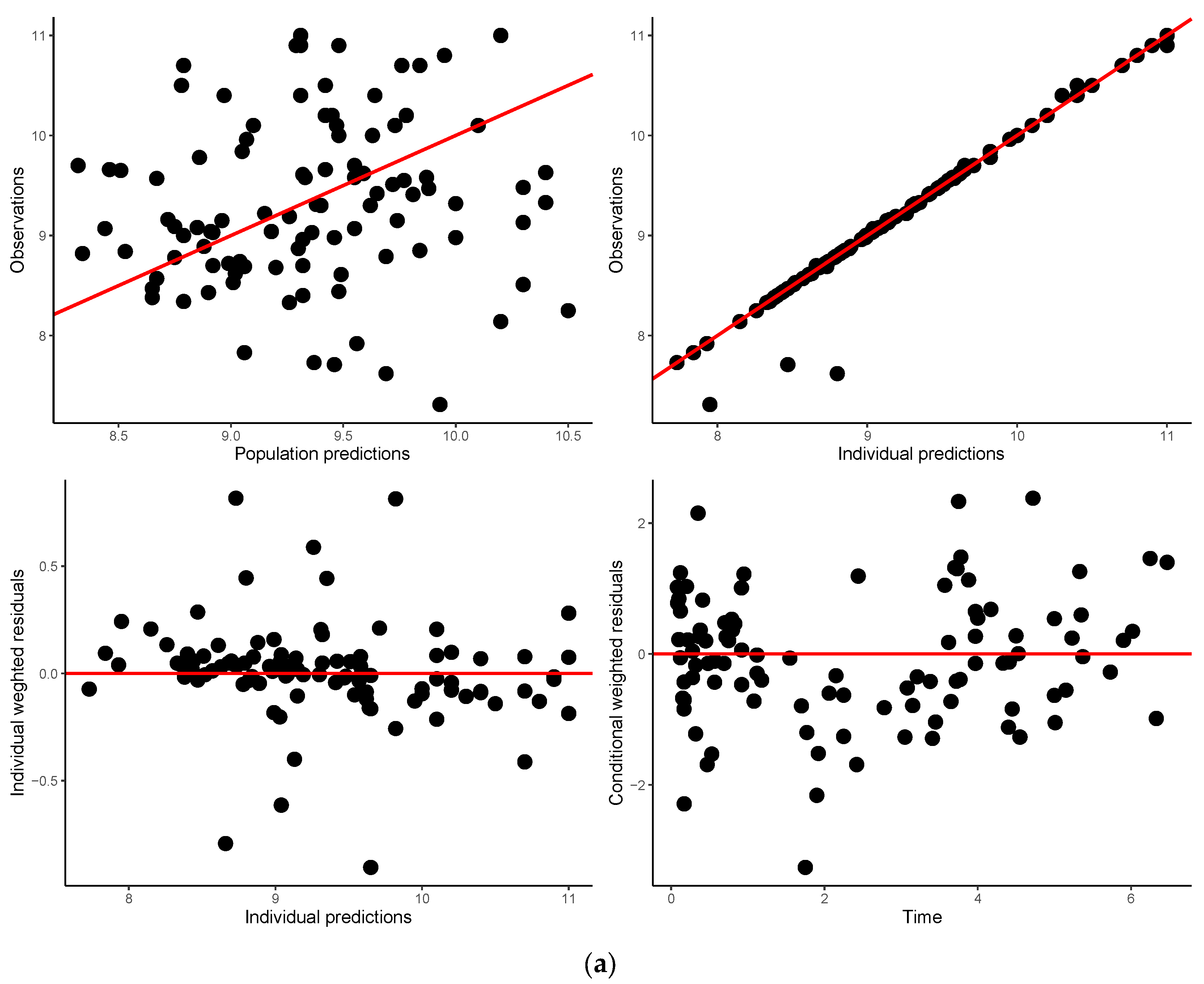
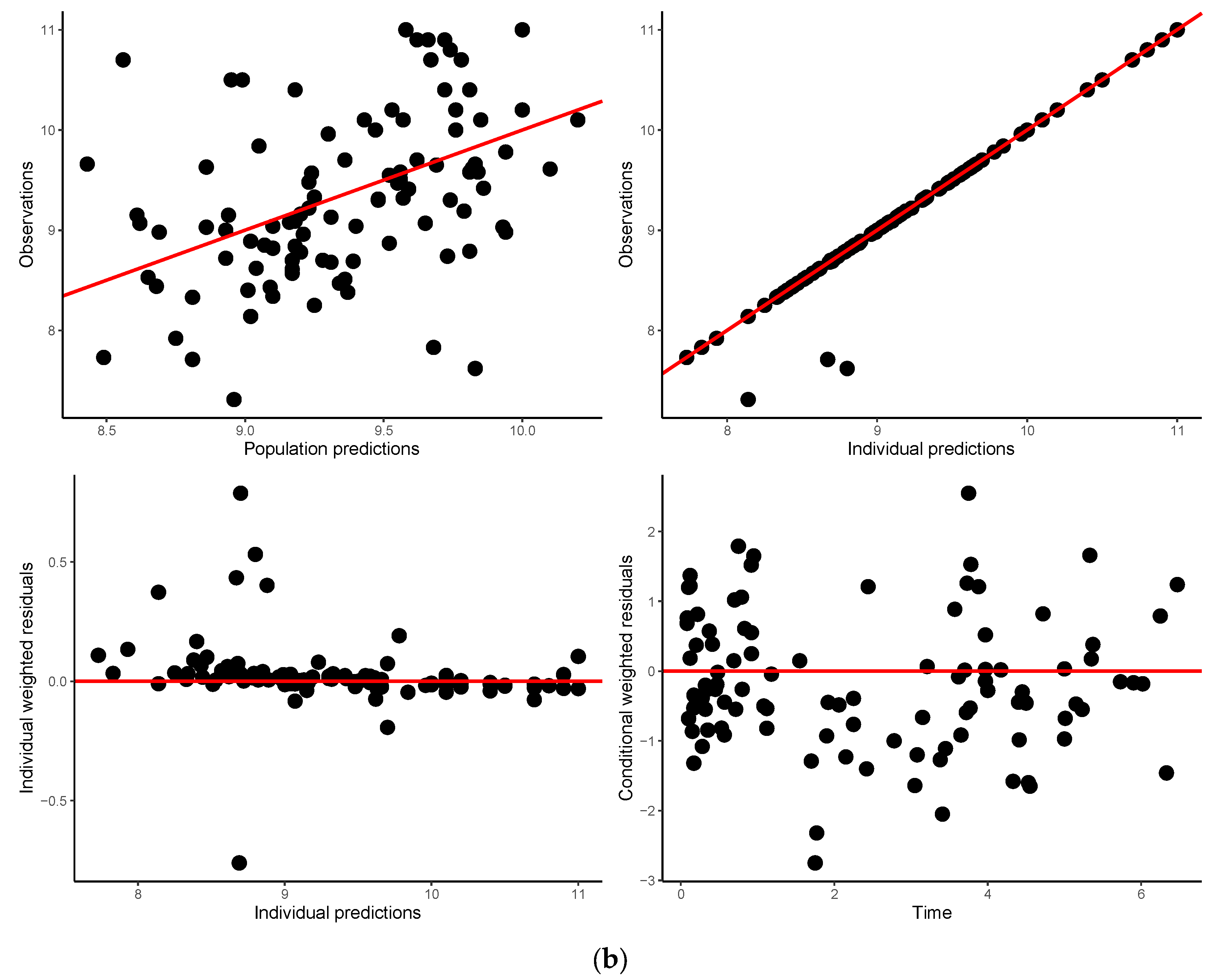
3.5. Model Validation
3.6. Model Application
4. Discussion
4.1. Gentamicin Population Pharmacokinetics
4.2. Application
4.3. Challenge
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chitty, K.M.; Chan, B.; Pulanco, C.L.; Luu, S.; Egunsola, O.; Buckley, N.A. Discontinuities and disruptions in drug dosage guidelines for the paediatric population. Br. J. Clin. Pharmacol. 2018, 84, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.; Selen, A.; Avant, D.; Chaurasia, C.; Crescenzi, T.; Gieser, G.; Di Giacinto, J.; Huang, S.M.; Lee, P.; Mathis, L.; et al. Improving pediatric dosing through pediatric initiatives: What we have learned. Pediatrics 2008, 121, 530–539. [Google Scholar] [CrossRef]
- Mahmood, I. Dosing in Children: A critical Review of the Pharmacokinetic Allometric Scaling and Modelling Approaches in Paediatric Drug Development and Clinical Settings. Clin. Pharmacokinet. 2014, 53, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Samant, T.; Mangal, N.; Lukacova, V.; Schmidt, S. Quantitative clinical pharmacology for size and age scaling in paediatric drug development: A systematic review. J. Clin. Pharmacol. 2015, 55, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Cimpello, L.; Khine, H.; Avner, J. Practice patterns of pediatric versus general emergency physicians for pain management of fractures in pediatric patients. Pediatr. Emerg. Care 2004, 20, 228–232. [Google Scholar] [CrossRef]
- Conroy, S.; Choonara, I.; Impicciatore, P.; Mohn, A.; Arnell, H.; Rane, A.; Knoeppel, C.; Seyberth, H.; Pandolfini, C.; Raffaelli, M.P.; et al. Survey of unlicensed and off label drug use in paediatric wards in European countries. Br. Med. J. 2000, 320, 79–82. [Google Scholar] [CrossRef]
- Cella, M.; Knibbe, C.; Danhof, M.; Della Pasqua, O. What is the right dose for children? Br. J. Clin. Pharmacol. 2010, 70, 597–603. [Google Scholar] [CrossRef]
- Hewitt, W. Gentamicin: Toxicity in perspective. Postgr. Med. J. 1974, 50 (Suppl. S7), 55–59. [Google Scholar]
- Gailiuanas, P.J.; Dominguez-Moreno, M.; Lazarus, J.; Lowrie, E.; Gottlieb, M.; Merrill, J. Vestibular toxicity of gentamicin; incidence in patients receiving longterm hemodialysis therapy. Arch. Intern. Med. 1978, 138, 1621–1624. [Google Scholar] [CrossRef]
- Dahlgren, J.; Anderson, E.; Hewitt, W. Gentamicin blood levels: A guide to nephrotoxicity. Antimicrob. Agents Chemother. 1975, 8, 58–62. [Google Scholar] [CrossRef]
- Assael, B.; Gianni, V.; Marini, A.; Peneff, P.; Sereni, F. Gentamicin dosage in preterm and term neonates. Arch. Dis. Child. 1977, 52, 883–886. [Google Scholar] [CrossRef]
- Morselli, P. Clinical pharmacology of the perinatal period and early infancy. Clin. Pharmacokinet. 1989, 17 (Suppl. S1), 13–28. [Google Scholar] [CrossRef]
- Rocha, M.J.; Almeida, A.M.; Afonso, E.; Martins, V.; Santos, J.; Leitao, F.; Falcão, A.C. The Kinetic Profile of Gentamicin in Premature Neonates. J. Pharm. Pharmacol. 2000, 52, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Faura, C.C.; Feret, M.A.; Horga, J.F. Monitoring Serum Levels of Gentamicin to Develop a New Regimen for Gentamicin Dosage in Newborns. Ther. Drug Monit. 1991, 13, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Austin, M.L.; Frye, R.F. Evaluation of gentamicin pharmacokinetics and dosing protocols in 195 neonates. Am. J. Health-Syst. Pharm. 1998, 55, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- McCracken, G.; Nelson, L. (Eds.) Aminoglycosides. In Antimicrobial Therapy for Newborns; Grune & Stratton, Inc.: New York, NY, USA, 1983; pp. 44–65. [Google Scholar]
- Mulhall, A.; de Louvois, J.; Hurley, R. Incidence of potentially toxic concentrations of gentamicin in the neonate. Arch. Dis. Child. 1983, 58, 897–900. [Google Scholar] [CrossRef]
- Edwards, C.; Low, D.; Bissenden, J. Gentamicin dosage for newborns. Lancet 1986, 1, 508–509. [Google Scholar] [CrossRef]
- Weber, W.; Kewitz, G.; Rost, K.L.; Looby, M.; Nitz, M.; Harnisch, L. Population kinetics of gentamicin in neonates. Eur. J. Clin. Pharmacol. 1993, 44 (Suppl. S1), 23–25. [Google Scholar] [CrossRef]
- Thomson, A.; Way, S.; Bryson, S.; McGovern, E.; Kelman, A. Population pharmacokinetics of gentamicin in neonates. Dev. Pharmacol. Ther. 1988, 11, 173–179. [Google Scholar]
- Metsvaht, T.; Pisarev, H.; Ilmoja, M.L.; Parm, Ü.; Maipuu, L.; Merila, M.; Müürsepp, P.; Lutsar, I. Clinical parameters predicting failure of empirical antibacterial therapy in early onset neonatal sepsis, identified by classification and regression tree analysis. BMC Pediatr. 2009, 9, 72. [Google Scholar] [CrossRef]
- Lingvall, M.; Reith, D.; Broadbent, R. The effect of sepsis upon gentamicin pharmacokinetics in neonates. Br. J. Clin. Pharmacol. 2005, 59, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Rayner, C.R.; Smith, P.F.; Andes, D.; Andrews, K.; Derendorf, H.; Friberg, L.E.; Hanna, D.; Lepak, A.; Mills, E.; Polasek, T.M.; et al. Model-Informed Drug Development for Anti-Infectives: State of the Art and Future. Clin. Pharmacol. Ther. 2021, 109, 867–891. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L.; Liu, W.; Fikes, S.; Cirz, R.; Robbins, N.; Kurhanewicz, S.; Rodriquez, J.; Brown, D.; Baluya, D.; Louie, A. Interaction of drug- and granulocyte-mediated killing of Pseudomonas aeruginosa in a murine pneumonia model. J. Infect. Dis. 2014, 210, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Thorsted, A.; Bouchene, S.; Tano, E.; Castegren, M.; Lipcsey, M.; Sjölin, J.; Karlsson, M.O.; Friberg, L.E.; Nielsen, E.I. A non-linear mixed effect model for innate immune response: In vivo kinetics of endotoxin and its induction of the cytokines tumor necrosis factor alpha and interleukin-6. PLoS ONE 2019, 14, e0211981. [Google Scholar] [CrossRef] [PubMed]
- Chacha, F.; Mirambo, M.M.; Mushi, M.F.; Kayange, N.; Zuechner, A.; Kidenya, B.R.; Mshana, S.E. Utility of qualitative C-reactive protein assay and white blood cells counts in the diagnosis of neonatal septicaemia at Bugando Medical Centre, Tanzania. BMC Pediatr. 2014, 14, 248. [Google Scholar] [CrossRef]
- Jones, C.D.; Sun, H.; Ette, E.I. Designing cross-sectional population pharmacokinetic studies: Implications for pediatric and animal studies. Clin. Res. Reg. Aff. 1996, 13, 133–165. [Google Scholar] [CrossRef]
- Vervelde, M.L.; Rademaker, C.M.; Krediet, T.G.; Fleer, A.; van Asten, P.; van Dijk, A. Population pharmacokinetics of gentamicin in preterm neonates: Evaluation of a once-daily dosage regimen. Ther. Drug Mon. 1999, 21, 514. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Almeida, A.; Falcao, A.; Caramona, M. Performance of gentamicin population kinetic parameters in Portuguese neonates. Pharm. World Sci. 2007, 29, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Hayani, K.C.; Hatzopoulos, F.K.; Frank, A.L.; Thummala, M.R.; Hantsch, M.J.; Schatz, B.M.; John, E.G.; Vidyasagar, D. Pharmacokinetics of once-daily dosing of gentamicin in neonates. J. Pediatr. 1997, 131, 76–80. [Google Scholar] [CrossRef]
- Ette, E.I.; Williams, P.; Sun, H.; Fadiran, E.; Ajayi, F.O.; Onyiah, L.C. The process of knowledge discovery from large pharmacokinetic data sets. J. Clin. Pharmacol. 2001, 41, 25–34. [Google Scholar] [CrossRef]
- Ette, E.I.; Williams, P.; Kim, Y.; Lane, J.; Liu, M.; Capparelli, E.V. Model Appropriateness and Population Pharmacokinetic Modeling. Clin. Pharmacol. 2013, 43, 610–623. [Google Scholar] [CrossRef]
- Germovsek, E.; Kent, A.; Metsvaht, T.; Lutsar, I.; Klein, N.; Turner, M.A.; Sharland, M.; Nielsen, E.I.; Heath, P.T.; Standing, J.F. Development and evaluation of a gentamicin pharmacokinetic model that facilitates opportunistic gentamicin therapeutic drug monitoring in neonates and infants. Antimicrob. Agents Chemother. 2016, 60, 4869–4877. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, C.M.; Kostan, E.; Broadbent, R.; Medlicott, N.; Reith, D. Evaluation of the effect of intravenous volume expanders upon the volume of distribution of gentamicin in septic neonates. Biopharm. Drug Dispos. 2009, 30, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Hussein, Z.; Pitsiu, M.; Majid, O.; Aarons, L.; De Longueville, M.; Stockis, A. Retrospective population pharmacokinetics of levocetirizine in atopic children receiving cetirizine: The ETAC® study. Br. J. Clin. Pharmacol. 2005, 59, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Adelman, M.; Evans, E.; Schentag, J. Two-compartment comparison of gentamicin and tobromaycin in normal volunteers. Antimicrob. Agents Chemother. 1982, 22, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Guidi, M.; Giannoni, E.; Werner, D.; Buclin, T.; Widmer, N.; Csajka, C. Population pharmacokinetic study of gentamicin in a large cohort of premature and term neonates. Br. J. Clin. Pharmacol. 2014, 78, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- García, B.; Barcia, E.; Pérez, F.; Molina, I.T. Population pharmacokinetics of gentamicin in premature newborns. J. Antimicrob. Chemother. 2006, 58, 372–379. [Google Scholar] [CrossRef]
- Kelman, A.W.; Thomson, A.H.; Whiting, B.; Bryson, S.M.; Steedman, D.A.; Mawer, G.E.; Samba-Donga, L.A. Estimation of gentamicin clearance and volume of distribution in neonates and young children. Br. J. Clin. Pharmacol. 1984, 18, 685–692. [Google Scholar] [CrossRef]
- Di Stefano, J.J., III. Optimized blood sampling protocols and sequential design of kinetic experiments. Am. J. Physiol. 1981, 240, R259–R296. [Google Scholar]
- Salerno, S.N.; Liao, Y.; Jackson, W.; Greenberg, R.G.; McKinzie, C.J.; McCallister, A.; Benjamin, D.K.; Laughon, M.M.; Sanderson, K.; Clark, R.H.; et al. Association between Nephrotoxic Drug Combinations and Acute Kidney Injury in the Neonatal Intensive Care Unit. J. Pediatr. 2021, 228, 213–219. [Google Scholar] [CrossRef]
- Jensen, P.; Edgren, B.; Brundage, R. Population Pharmacokinetics of Gentamicin in Neonates Using a Nonlinear, Mixed-Effects Model. Pharmacotherapy 1992, 12, 178–182. [Google Scholar] [CrossRef]
- Botha, J.H.; Du Preez, M.J.; Adhikari, M. Population pharmacokinetics of gentamicin in South African newborns. Eur. J. Clin. Pharmacol. 2003, 59, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.H.; Kokwaro, G.O.; Muchohi, S.N.; English, M.; Mohammed, S.; Edwards, G. Population pharmacokinetics of intramuscular gentamicin administered to young infants with suspected severe sepsis in Kenya. Br. J. Clin. Pharmacol. 2003, 56, 25–31. [Google Scholar] [CrossRef]
- Ernandez, T.; Mayadas, T. The changing landscape of renal inflammation. Trends Mol. Med. 2016, 22, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Sorsa, A. Diagnostic significance of white blood cell count and c-reactive protein in neonatal sepsis; Asella Referral Hospital, South East Ethiopia. Open Microbiol. J. 2018, 12, 209. [Google Scholar] [CrossRef]
- Caldas, J.P.; Marba, S.T.; Blotta, M.H.; Calil, R.; Morais, S.S.; Oliveira, R.T. Accuracy of white blood cell count, C-reactive protein, interleukin-6 and tumor necrosis factor alpha for diagnosing late neonatal sepsis. J. Pediatr. 2008, 84, 536–542. [Google Scholar] [CrossRef]
- Darmstadt, G.L.; Miller-Bell, M.; Batra, M.; Law, P.; Law, K. Extended-interval dosing of gentamicin for treatment of neonatal sepsis in developed and developing countries. J. Health Popul. Nutr. 2008, 26, 163–182. [Google Scholar]
- Perlman, J.M.; McMenamin, J.B.; Volpe, J.J. Fluctuating cerebral blood flow in RDS; relationship to the development of intraventricular hemorrhage. N. Engl. J. Med. 1983, 309, 204–209. [Google Scholar] [CrossRef]
- Kauffman, R.E.; Kearns, G.L. Pharmacokinetic studies in pediatric patients: Clinical and ethical considerations. Clin. Pharmacokinet. 1992, 23, 10–29. [Google Scholar] [CrossRef]
- Al-Banna, M.K.; Kelman, A.W.; Whiting, B. Experimental design and efficient parameter estimation in population pharmacokinetics. J. Pharmacokinet. Biopharm. 1990, 18, 347–360. [Google Scholar] [CrossRef]
- D’Argenio, D.Z. Optimal sampling times for pharmacokinetic experiments. J. Pharmacokinet. Biopharm. 1981, 6, 41–53. [Google Scholar] [CrossRef]
- Ette, E.I.; Sun, H.; Ludden, T.M. Design of population pharmacokinetic studies. Proc. Am. Stat. Assoc. 1994, 487–492. [Google Scholar]
- Fjalstad, J.W.; Laukli, E.; Van Den Anker, J.N.; Klingenberg, C. High-dose gentamicin in newborn infants: Is it safe? Eur. J. Pediatr. 2014, 173, 489–495. [Google Scholar] [CrossRef] [PubMed]
- British Medical Association; Royal College of Paediatrics and Child Health RPS of GB. BNF for Children; RCPCH Publications; BMJ Publishing Group, RPS Publishing: London, UK, 2022. [Google Scholar]
- Thomson Reuters Clinical Editorial Staff. Neofax 2011, 24th ed.; PDR Network: Hanover, NJ, USA, 2011. [Google Scholar]
- Pillay, S.; Horn, A. (Eds.) Neonatal Guidelines and Drug Doses, 6th ed.; Neonatal Guidelines: Cape Town, South Africa, 2022. [Google Scholar]
- Valitalo, P.A.; van den Anker, J.N.; Allegaert, K.; de Cock, R.F.; de Hoog, M.; Simons, S.H.; Mouton, J.W.; Knibbe, C.A. Novel model-based dosing guidelines for gentamicin and tobramycin in preterm and term neonates. J. Antimicrob. Chemother. 2015, 70, 2074–2077. [Google Scholar] [CrossRef]
- Bijleveld, Y.A.; Van Den Heuvel, M.E.; Hodiamont, C.J.; Mathôt, R.A.A.; De Haan, T.R. Population pharmacokinetics and dosing considerations for gentamicin in newborns with suspected or proven sepsis caused by gram-negative bacteria. Antimicrob. Agents Chemother. 2017, 61, e01304-16. [Google Scholar] [CrossRef]
- Hossain, M.M.; Chowdhury, N.A.; Shirin, M.; Saha, S.K.; Miller-Bell, M.; Edwards, D.; Aranda, J.; Coffey, P.; Darmstadt, G.L. Simplified dosing of gentamicin for treatment of sepsis in bangladeshi neonates. J. Health Popul. Nutr. 2009, 27, 640–645. [Google Scholar] [PubMed]
- McDermott, J.H.; Mahaveer, A.; James, R.A.; Booth, N.; Turner, M.; Harvey, K.E.; Miele, G.; Beaman, G.M.; Stoddard, D.C.; Tricker, K.; et al. Rapid point-of-care genotyping to avoid aminoglycoside-induced ototoxicity in neonatal intensive care. JAMA Pediatr. 2022, 176, 486–492. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | n (%) |
|---|---|
| Females | 23 (44.2) |
| Males | 29 (55.8) |
| Preterm (<37 weeks GA) | 43 (82.7) |
| Total number of neonates | 52 |
| Median (range) | |
| Birth weight (kg) | 1.57 (0.90–3.92) |
| Gestational age (weeks) | 32 (24–40) |
| Postnatal age (days) | 4.0 (1.0–17) |
| Height (cm) | 41 (30–53) |
| White blood cell count (×109/L) | 11.0 (1.67–37.4) |
| Serum creatinine (mg/dL) | 0.72 (0.20–1.66) |
| Gentamicin dose (mg) | 7.9 (4.0–17) |
| Run Number | Description | cf | OFV | LLD | df | LRT * Significant |
|---|---|---|---|---|---|---|
| 1 | Base Model: CLRef | |||||
| VRef | - | 15.076 | ||||
| 2 | CL~WT | 1 | 9.903 | −5.173 | 1 | yes |
| 3 | CL~PNA | 1 | 11.499 | −3.577 | 1 | no |
| 4 | CL~FMAT | 1 | 1.802 | −13.274 | 2 | yes |
| 5 | CL~WBC | 1 | 0.653 | −14.423 | 1 | yes |
| 6 | Cl~RSCR | 1 | 12.543 | −2.533 | 1 | no |
| 7 | V~WBC | 1 | 14.989 | −0.087 | 1 | no |
| 8 | V~WT | 1 | −7.403 | −22.479 | 1 | yes |
| 9 | CL~WBC, FMAT | 4 | −2.346 | −4.148 | 1 | yes |
| 10 | CL~WBC, FMAT, WT | 9 | −8.675 | −6.329 | 1 | yes |
| 11 | CL~WBC, FMAT, WT | 10 | −22.151 | −13.476 | 1 | yes |
| V~WT |
| Parameter | Original Estimate | 90% Bootstrap Confidence Interval | Shrinkage (%) |
|---|---|---|---|
| CL (L/h) | 0.196 | 0.132, 0.228 | |
| V (L) | 0.417 | 0.330, 0.476 | |
| V_WT | 1.76 | 1.39, 2.34 | |
| CL_WT | 1.30 | 0.558, 1.68 | |
| CL_WBC | −0.560 | −1.70, 0.375 | |
| GAMMA | 0.551 | Fixed | |
| PNA50 (yr) | 0.0332 | Fixed | |
| a Intersubject variability | |||
| 1.00 × 10−5 (0.31%) | −0.289, 0.289 | 3.19 | |
| 0.00024 | −0.221, 0.234 | ||
| 0.501 (70.8%) | 0.436, 0.679 | 3.18 | |
| 6.23 (250%) | 9.10, 13.4 | 1.26 | |
| b Residual variability | |||
| ResADDITIVE1 | 4.96 | 2.99, 16.83 | 9.37 |
| ResPROPORTIONAL1 | −0.99 | −1.235, −0.745 | |
| ResADDITIVE2 | 1.00 × 10−5 | −0.00017, 0.00019 | |
| ResPROPORTIONAL2 | 0.0205 | −1.95, 1.96 | |
| Concentration Percentile | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmin (<2 mg/L) | Cmax (5–12 mg/L) | Concentration above 12 mg/L | |||||||
| 5th | 50th | 95th | 5th | 50th | 95th | 5th | 50th | 95th | |
| Postnatal age (days) | |||||||||
| 1–2 | 0.52 | 1.30 | 1.94 | 5.27 | 7.28 | 10.30 | 12.10 | 12.80 | 14.66 |
| 3–5 | 0.43 | 1.26 | 1.93 | 5.12 | 6.89 | 10.10 | 12.10 | 12.60 | 14.00 |
| 6–10 | 0.46 | 1.26 | 1.93 | 5.07 | 6.56 | 9.88 | 12.23 | 12.40 | 13.97 |
| >10 | 0.42 | 1.21 | 1.88 | 5.06 | 6.55 | 9.61 | 12.70 | 12.70 | 12.70 |
| Weight (g) | |||||||||
| <1000 | 0.42 | 1.18 | 1.88 | 5.09 | 6.96 | 10.10 | 12.14 | 12.40 | 13.62 |
| 1000–1499 | 0.41 | 1.25 | 1.93 | 5.11 | 6.83 | 10.00 | 12.10 | 12.70 | 14.90 |
| 1500–2499 | 0.45 | 1.26 | 1.91 | 5.13 | 6.90 | 10.20 | 12.10 | 12.70 | 14.00 |
| >2500 | 0.53 | 1.40 | 1.94 | 5.23 | 7.22 | 10.20 | 12.10 | 12.70 | 14.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singu, B.S.; Verbeeck, R.K.; Pieper, C.H.; Ette, E.I. Confirming the Suitability of a Gentamicin Dosing Strategy in Neonates Using the Population Pharmacokinetic Approach with Truncated Sampling Duration. Children 2024, 11, 898. https://doi.org/10.3390/children11080898
Singu BS, Verbeeck RK, Pieper CH, Ette EI. Confirming the Suitability of a Gentamicin Dosing Strategy in Neonates Using the Population Pharmacokinetic Approach with Truncated Sampling Duration. Children. 2024; 11(8):898. https://doi.org/10.3390/children11080898
Chicago/Turabian StyleSingu, Bonifasius Siyuka, Roger Karel Verbeeck, Clarissa Hildegard Pieper, and Ene I. Ette. 2024. "Confirming the Suitability of a Gentamicin Dosing Strategy in Neonates Using the Population Pharmacokinetic Approach with Truncated Sampling Duration" Children 11, no. 8: 898. https://doi.org/10.3390/children11080898