



Genome-Wide Mapping of Consanguineous Families Confirms Previously Implicated Gene Loci and Suggests New Loci in Specific Language Impairment (SLI)
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Individuals
2.2. SNP Genotyping and Linkage Analysis
2.3. Homozygosity Mapping
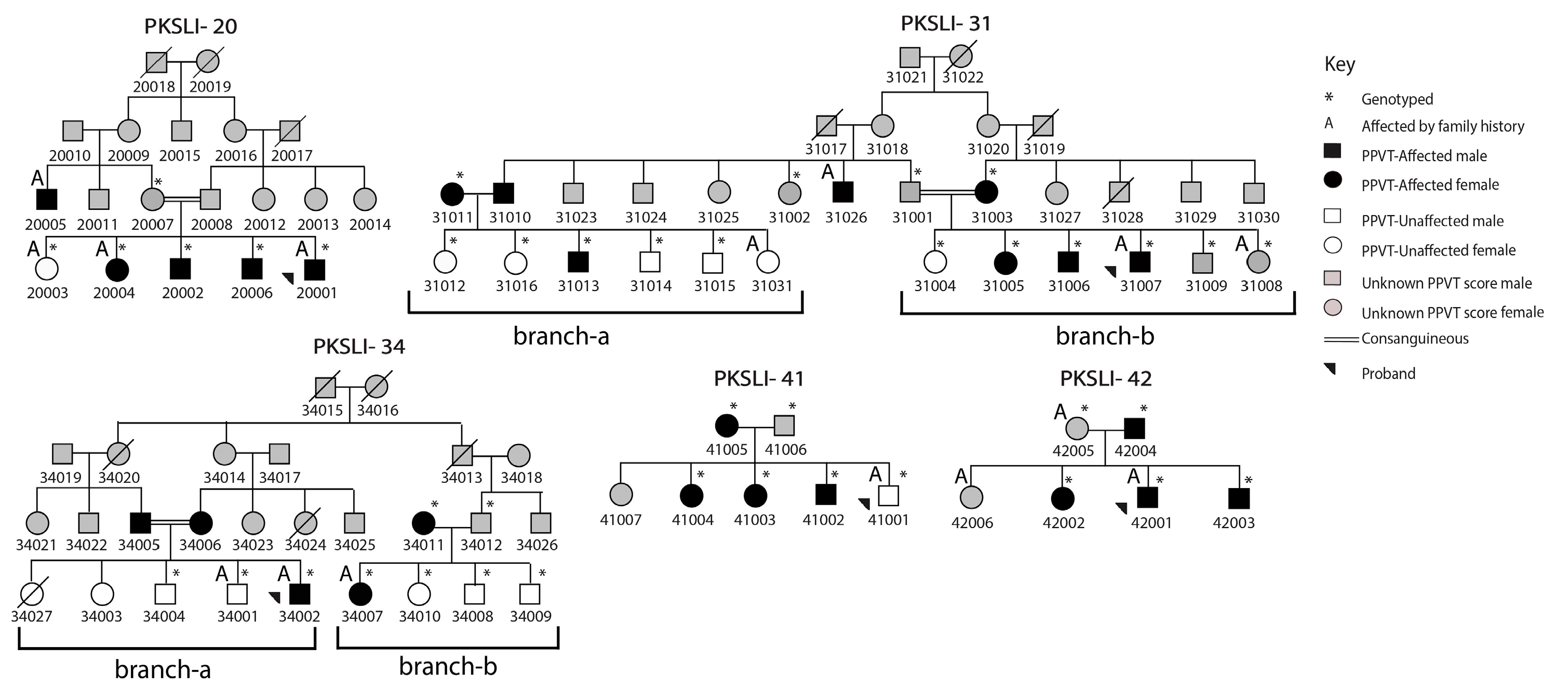
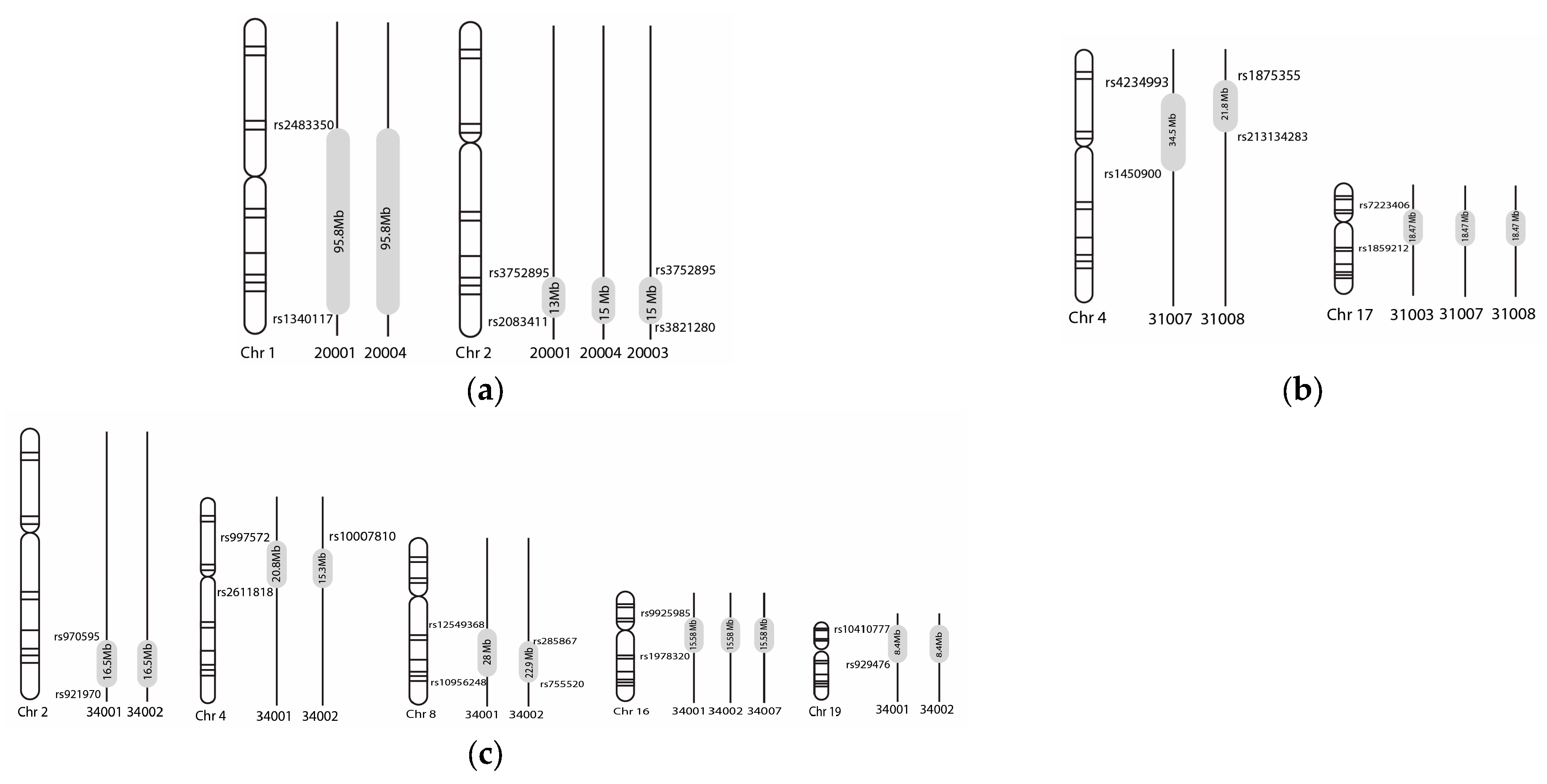
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonard, L.B. Children with Specific Language Impairment; MIT Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Norbury, C.F.; Gooch, D.; Wray, C.; Baird, G.; Charman, T.; Simonoff, E.; Vamvakas, G.; Pickles, A. The impact of nonverbal ability on prevalence and clinical presentation of language disorder: Evidence from a population study. J. Child Psychol. Psychiatry 2016, 57, 1247–1257. [Google Scholar] [CrossRef]
- Young, A.R.; Beitchman, J.H.; Johnson, C.; Douglas, L.; Atkinson, L.; Escobar, M.; Wilson, B. Young adult academic outcomes in a longitudinal sample of early identified language impaired and control children. J. Child Psychol. Psychiatry 2002, 43, 635–645. [Google Scholar] [CrossRef]
- Tomblin, J.B. Educational and psychosocial outcomes of language impairment in kindergarten. In Understanding Individual Differences in Language Development across the School Years; Psychology Press: London, UK, 2014; pp. 166–203. [Google Scholar]
- Rice, M.L.; Smith, S.D.; Gayán, J. Convergent genetic linkage and associations to language, speech and reading measures in families of probands with specific language impairment. J. Neurodev. Disord. 2009, 1, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Wankoff, L.S. Warning signs in the development of speech, language, and communication: When to refer to a speech-language pathologist. J. Child Adolesc. Psychiatr. Nurs. 2011, 24, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Roth, F.P. Guiding principles and clinical applications for speech-language pathology practice in early intervention. Lang. Speech Hear. Serv. Sch. 2011, 42, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kalnak, N.; Peyrard-Janvid, M.; Sahlén, B.; Forssberg, H. Family history interview of a broad phenotype in specific language impairment and matched controls. Genes Brain Behav. 2012, 11, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Sharp, H.M.; Hillenbrand, K. Speech and language development and disorders in children. Pediatr. Clin. N. Am. 2008, 55, 1159–1173. [Google Scholar] [CrossRef]
- Betz, S.K.; Eickhoff, J.R.; Sullivan, S.F. Factors influencing the selection of standardized tests for the diagnosis of specific language impairment. Lang. Speech Hear. Serv. Sch. 2013, 44, 133–146. [Google Scholar] [CrossRef]
- Bishop, D.V.; North, T.; Donlan, C. Genetic basis of specific language impairment: Evidence from a twin study. Dev. Med. Child Neurol. 1995, 37, 56–71. [Google Scholar] [CrossRef]
- Bishop, D.V. The role of genes in the etiology of specific language impairment. J. Commun. Disord. 2002, 35, 311–328. [Google Scholar] [CrossRef]
- Marchini, J.; Donnelly, P.; Cardon, L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005, 37, 413–417. [Google Scholar] [CrossRef]
- Hu, H.; Kahrizi, K.; Musante, L.; Fattahi, Z.; Herwig, R.; Hosseini, M.; Oppitz, C.; Abedini, S.S.; Suckow, V.; Larti, F. Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 2019, 24, 1027–1039. [Google Scholar] [CrossRef]
- Fahiminiya, S.; Almuriekhi, M.; Nawaz, Z.; Staffa, A.; Lepage, P.; Ali, R.; Hashim, L.; Schwartzentruber, J.; Abu Khadija, K.; Zaineddin, S. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin. Genet. 2014, 86, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Vieland, V.J.; Bassett, A.S.; Tallal, P.; Brzustowicz, L.M. A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet. 2002, 71, 45–55. [Google Scholar] [CrossRef]
- Reader, R.H.; Covill, L.E.; Nudel, R.; Newbury, D.F. Genome-wide studies of specific language impairment. Curr. Behav. Neurosci. Rep. 2014, 1, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Consortium, S. A genomewide scan identifies two novel loci involved in specific language impairment. Am. J. Hum. Genet. 2002, 70, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, P.; Newbury, D.F.; Jara, L.; De Barbieri, Z.; Mirza, G.; Palomino, H.M.; Fernández, M.A.; Cazier, J.-B.; Monaco, A.P.; Palomino, H. Genome-wide analysis of genetic susceptibility to language impairment in an isolated Chilean population. Eur. J. Hum. Genet. 2011, 19, 687–695. [Google Scholar] [CrossRef]
- Villanueva, P.; Nudel, R.; Hoischen, A.; Fernández, M.A.; Simpson, N.H.; Gilissen, C.; Reader, R.H.; Jara, L.; Echeverry, M.M.; Francks, C.; et al. Exome Sequencing in an Admixed Isolated Population Indicates NFXL1 Variants Confer a Risk for Specific Language Impairment. PLoS Genet. 2015, 11, e1005336. [Google Scholar] [CrossRef]
- Andres, E.M.; Earnest, K.K.; Zhong, C.; Rice, M.L.; Raza, M.H. Family-based whole-exome analysis of specific language impairment (SLI) identifies rare variants in BUD13, a component of the retention and splicing (RES) complex. Brain Sci. 2021, 12, 47. [Google Scholar] [CrossRef]
- Andres, E.M.; Hafeez, H.; Yousaf, A.; Riazuddin, S.; Rice, M.L.; Basra, M.A.R.; Raza, M.H. A genome-wide analysis in consanguineous families reveals new chromosomal loci in specific language impairment (SLI). Eur. J. Hum. Genet. 2019, 27, 1274–1285. [Google Scholar] [CrossRef]
- Raza, M.H.; Amjad, R.; Riazuddin, S.; Drayna, D. Studies in a consanguineous family reveal a novel locus for stuttering on chromosome 16q. Hum. Genet. 2012, 131, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Gertz, E.M.; Mundorff, J.; Lukong, J.; Kuster, J.; Schäffer, A.A.; Drayna, D. Linkage analysis of a large African family segregating stuttering suggests polygenic inheritance. Hum. Genet. 2013, 132, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.; Wang, J.; Leal, S.M. Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 2015, 16, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.E.; Vargha-Khadem, F.; Watkins, K.E.; Monaco, A.P.; Pembrey, M.E. Localisation of a gene implicated in a severe speech and language disorder. Nat. Genet. 1998, 18, 168–170. [Google Scholar] [CrossRef]
- Terwilliger, J.D.; Ott, J. Handbook of Human Genetic Linkage; JHU Press: Baltimore, MD, USA, 1994. [Google Scholar]
- Seelow, D.; Schuelke, M.; Hildebrandt, F.; Nürnberg, P. HomozygosityMapper—An interactive approach to homozygosity mapping. Nucleic Acids Res. 2009, 37, W593–W599. [Google Scholar] [CrossRef]
- Consortium, S. Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am. J. Hum. Genet. 2004, 74, 1225–1238. [Google Scholar]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Smith, B.J.; Vieland, V.J.; Tallal, P.; Brzustowicz, L.M. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered. 2004, 57, 10–20. [Google Scholar] [CrossRef]
- Truong, D.; Shriberg, L.; Smith, S.; Chapman, K.; Scheer-Cohen, A.; DeMille, M.; Adams, A.; Nato, A.; Wijsman, E.; Eicher, J.; et al. Multipoint genome-wide linkage scan for nonword repetition in a multigenerational family further supports chromosome 13q as a locus for verbal trait disorders. Hum. Genet. 2016, 135, 1329–1341. [Google Scholar]
- Nudel, R.; Simpson, N.H.; Baird, G.; O’Hare, A.; Conti-Ramsden, G.; Bolton, P.F.; Hennessy, E.R.; Consortium, S.; Ring, S.; Davey Smith, G.; et al. Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment. Genes Brain Behav. 2014, 13, 418–429. [Google Scholar] [CrossRef]
- Andres, E.M.; Earnest, K.K.; Smith, S.D.; Rice, M.L.; Raza, M.H. Pedigree-based gene mapping supports previous loci and reveals novel suggestive loci in specific language impairment. J. Speech Lang. Hear. Res. 2020, 63, 4046–4061. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, W.; Ying, D.; Cherny, S.S.; Hildebrandt, F.; Sham, P.C.; Lau, Y.L. Homozygosity mapping on a single patient—Identification of homozygous regions of recent common ancestry by using population data. Hum. Mutat. 2011, 32, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, T.; Andres, E.M.; Ashraf, K.; Basra, M.A.R.; Raza, M.H. Genome-wide analysis of runs of homozygosity in Pakistani controls with no history of speech or language-related developmental phenotypes. Ann. Hum. Biol. 2023, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.M.; Dunn, D.M. PPVT-4: Peabody Picture Vocabulary Test; Pearson Assessments: San Antonio, TX, USA; NCS Pearson, Inc.: Bloomington, MN, USA, 2007. [Google Scholar]
- Andres, E.M.; Neely, H.; Hafeez, H.; Yasmin, T.; Kausar, F.; Basra, M.A.R.; Raza, M.H. Study of rare genetic variants in TM4SF20, NFXL1, CNTNAP2, and ATP2C2 in Pakistani probands and families with language impairment. Meta Gene 2021, 30, 100966. [Google Scholar] [CrossRef]
- Chaudhry, I.S.; Rahman, S. The impact of gender inequality in education on rural poverty in Pakistan: An empirical analysis. Eur. J. Econ. Financ. Adm. Sci. 2009, 15, 174–188. [Google Scholar]
- Silberstein, M.; Weissbrod, O.; Otten, L.; Tzemach, A.; Anisenia, A.; Shtark, O.; Tuberg, D.; Galfrin, E.; Gannon, I.; Shalata, A.; et al. A system for exact and approximate genetic linkage analysis of SNP data in large pedigrees. Bioinformatics 2012, 29, 197–205. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Abreu, P.; Hodge, S.E. The power to detect linkage in complex disease by means of simple LOD-score analyses. Am. J. Hum. Genet. 1998, 63, 870–879. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Hunter, J.V.; Hanchard, N.A.; Willer, J.R.; Shaw, C.; Tian, Q.; Illner, A.; Wang, X.; Cheung, S.W.; Patel, A.; et al. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet. 2013, 93, 197–210. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Family/Branch | Chromosome | Linkage Loci (hg 19) * | Snip ID | Maximum LOD Score | Mode of Inheritance |
|---|---|---|---|---|---|
| PKSLI-20 | 6 | 6p21.1-p12.3 | rs736794 | 1.92 | recessive |
| PKSLI-31 | 12 | 12p11.22-q11.21 | rs581642 | 2.49 | dominant |
| PKSLI-31-a | 12 | 12q13.11-q13.12 | rs581642 | 1.87 | dominant |
| PKSLI-31 | 7 | 7q35-q36.1 | rs6464094 | 2 | recessive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousaf, A.; Hafeez, H.; Basra, M.A.R.; Rice, M.L.; Raza, M.H.; Shabbir, M.I. Genome-Wide Mapping of Consanguineous Families Confirms Previously Implicated Gene Loci and Suggests New Loci in Specific Language Impairment (SLI). Children 2024, 11, 1063. https://doi.org/10.3390/children11091063
Yousaf A, Hafeez H, Basra MAR, Rice ML, Raza MH, Shabbir MI. Genome-Wide Mapping of Consanguineous Families Confirms Previously Implicated Gene Loci and Suggests New Loci in Specific Language Impairment (SLI). Children. 2024; 11(9):1063. https://doi.org/10.3390/children11091063
Chicago/Turabian StyleYousaf, Adnan, Huma Hafeez, Muhammad Asim Raza Basra, Mabel L. Rice, Muhammad Hashim Raza, and Muhammad Imran Shabbir. 2024. "Genome-Wide Mapping of Consanguineous Families Confirms Previously Implicated Gene Loci and Suggests New Loci in Specific Language Impairment (SLI)" Children 11, no. 9: 1063. https://doi.org/10.3390/children11091063
APA StyleYousaf, A., Hafeez, H., Basra, M. A. R., Rice, M. L., Raza, M. H., & Shabbir, M. I. (2024). Genome-Wide Mapping of Consanguineous Families Confirms Previously Implicated Gene Loci and Suggests New Loci in Specific Language Impairment (SLI). Children, 11(9), 1063. https://doi.org/10.3390/children11091063