
Respiratory Support Strategies for Surgical Neonates: A Review


{kind=link}
{kind=link}
Abstract
:1. Introduction
Principles of Respiratory Support in the Surgical Neonate
2. Oesophageal Atresia-Tracheoesophageal Fistula
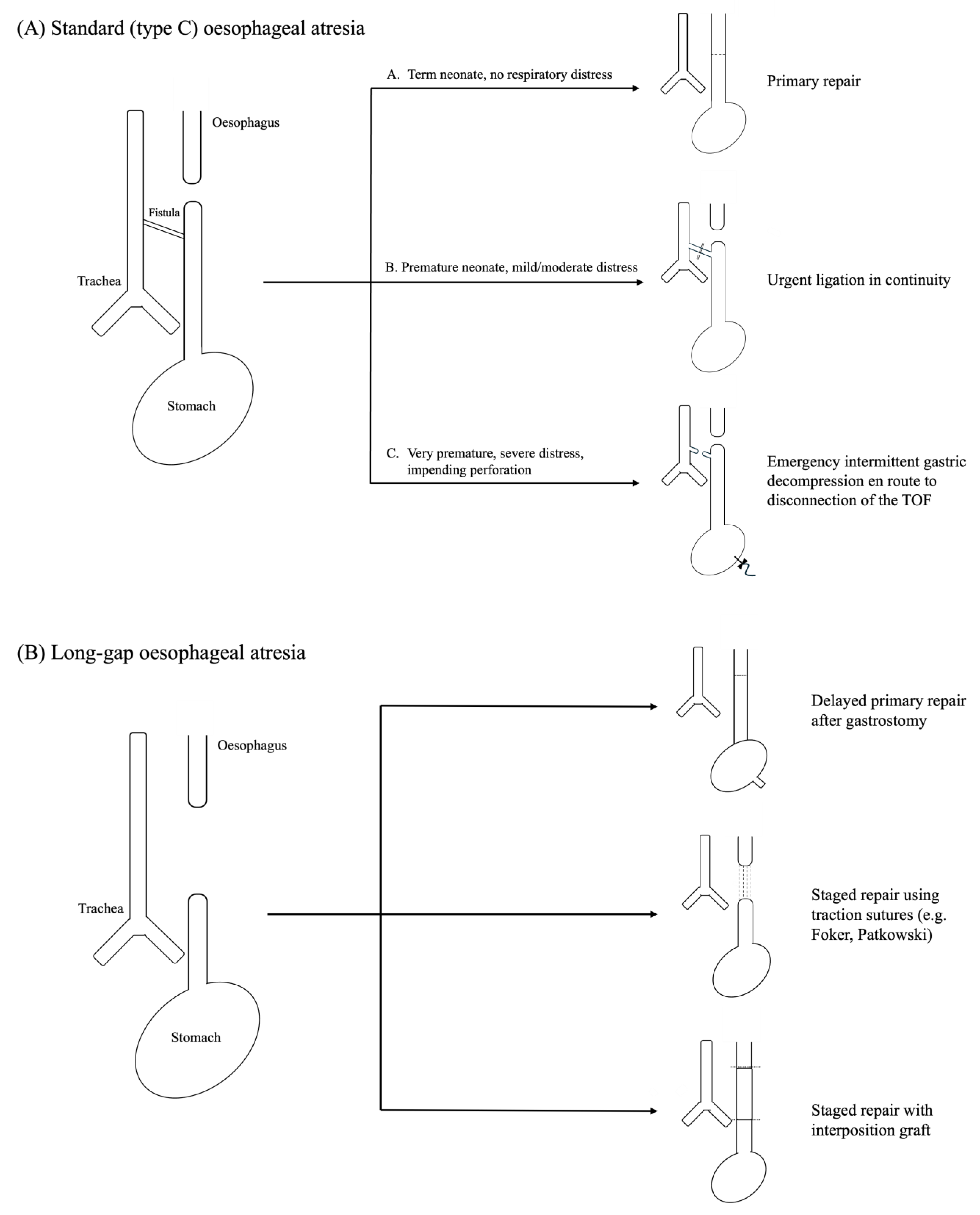
2.1. Preoperative Optimisation
2.2. Intraoperative Considerations
2.3. Postoperative Support
3. Congenital Diaphragmatic Hernia
3.1. Antenatal Management
3.2. Perinatal Stabilisation
3.3. Preoperative Respiratory Support
3.4. Perioperative Considerations
3.5. Postoperative Respiratory Support
4. Congenital Lung Malformations
4.1. Antenatal and Preoperative Support
4.2. Respiratory Support Strategies in Surgery for CLMs
4.3. Respiratory Considerations in Asymptomatic CLM Infants
5. Anterior Abdominal Wall Defects
5.1. Causes of Respiratory Distress in Infants with AWDs
5.2. Respiratory Considerations in Surgical Treatment of AWDs
5.3. Respiratory Complications Following Treatment of AWDs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kiger, J. Neonatal ventilation. Semin. Pediatr. Surg. 2022, 31, 151199. [Google Scholar] [CrossRef] [PubMed]
- Ruzic, A. Contemporary ventilatory strategies for surgical patients. Semin. Pediatr. Surg. 2019, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kaltsogianni, O.; Dassios, T.; Greenough, A. Neonatal respiratory support strategies-short and long-term respiratory outcomes. Front. Pediatr. 2023, 11, 1212074. [Google Scholar] [CrossRef] [PubMed]
- Course, C.W.; Kotecha, E.A.; Course, K.; Kotecha, S. The respiratory consequences of preterm birth: From infancy to adulthood. Br. J. Hosp. Med. 2024, 85, 1–11. [Google Scholar] [CrossRef]
- Narayanan, M.; Owers-Bradley, J.; Beardsmore, C.S.; Mada, M.; Ball, I.; Garipov, R.; Panesar, K.S.; Kuehni, C.E.; Spycher, B.D.; Williams, S.E.; et al. Alveolarization continues during childhood and adolescence: New evidence from helium-3 magnetic resonance. Am. J. Respir. Crit. Care Med. 2012, 185, 186–191. [Google Scholar] [CrossRef]
- Broemling, N.; Campbell, F. Anesthetic management of congenital tracheoesophageal fistula. Paediatr. Anaesth. 2011, 21, 1092–1099. [Google Scholar] [CrossRef]
- Hunt, R.W.; Perkins, E.J.; King, S. Peri-operative management of neonates with oesophageal atresia and tracheo-oesophageal fistula. Paediatr. Respir. Rev. 2016, 19, 3–9. [Google Scholar] [CrossRef]
- Nakayama, D.K. The history of surgery for esophageal atresia. J. Pediatr. Surg. 2020, 55, 1414–1419. [Google Scholar] [CrossRef]
- Bell, J.C.; Baynam, G.; Bergman, J.E.H.; Bermejo-Sánchez, E.; Botto, L.D.; Canfield, M.A.; Dastgiri, S.; Gatt, M.; Groisman, B.; Hurtado-Villa, P.; et al. Survival of infants born with esophageal atresia among 24 international birth defects surveillance programs. Birth Defects Res. 2021, 113, 945–957. [Google Scholar] [CrossRef]
- Spitz, L.; Kiely, E.M.; Morecroft, J.A.; Drake, D.P. Oesophageal atresia: At-risk groups for the 1990s. J. Pediatr. Surg. 1994, 29, 723–725. [Google Scholar] [CrossRef]
- Lopez, P.J.; Keys, C.; Pierro, A.; Drake, D.P.; Kiely, E.M.; Curry, J.I.; Spitz, L. Oesophageal atresia: Improved outcome in high-risk groups? J. Pediatr. Surg. 2006, 41, 331–334. [Google Scholar] [CrossRef] [PubMed]
- van Lennep, M.; Singendonk, M.M.J.; Dall’Oglio, L.; Gottrand, F.; Krishnan, U.; Terheggen-Lagro, S.W.J.; Omari, T.I.; Benninga, M.A.; van Wijk, M.P. Oesophageal atresia. Nat. Rev. Dis. Primers 2019, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D. VACTERL/VATER Association. Orphanet. J. Rare Dis. 2011, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Choumanovai, I.; Sanusi, A.; Evans, F. Anaesthetic Management of Tracheo-Oesophageal Fistula/Oesophageal Atresia. Available online: https://resources.wfsahq.org/atotw/anaesthetic-management-of-tracheo-oesophageal-fistula-oesophageal-atresia/ (accessed on 23 January 2025).
- Adebo, O.A. Oesophageal atresia and tracheo-oesophageal fistula: Review of a 10-year personal experience. West Afr. J. Med. 1990, 9, 164–169. [Google Scholar] [PubMed]
- Richenbacher, W.E.; Ballantine, T.V. Esophageal atresia, distal tracheoesophageal fistula, and an air shunt that compromised mechanical ventilation. J. Pediatr. Surg. 1990, 25, 1216–1218. [Google Scholar] [CrossRef]
- Templeton, J.M., Jr.; Templeton, J.J.; Schnaufer, L.; Bishop, H.C.; Ziegler, M.M.; O’Neill, J.A., Jr. Management of esophageal atresia and tracheoesophageal fistula in the neonate with severe respiratory distress syndrome. J. Pediatr. Surg. 1985, 20, 394–397. [Google Scholar] [CrossRef]
- van den Berg, J.; Johansen, M.; Disma, N.; Engelhardt, T.; Hansen, T.G.; Veyckemans, F.; Zielinska, M.; de Graaff, J.C. Perioperative anaesthetic management and short-term outcome of neonatal repair of oesophageal atresia with or without tracheo-oesophageal fistula in Europe: A sub-analysis of the neonate and children audit of anaesthesia practice in Europe (NECTARINE) prospective multicenter observational study. Eur. J. Anaesthesiol. 2023, 40, 936–945. [Google Scholar] [CrossRef]
- Li, S.; Cao, G.; Zhou, R.; Zhang, X.; Zhou, Y.; Tang, S.T. Feasible techniques in robotic thoracoscopic repair of congenital esophageal atresia: Case report and literature review. Surg. Case Rep. 2021, 7, 142. [Google Scholar] [CrossRef]
- Penikis, A.B.; Sescleifer, A.M.; Kunisaki, S.M. Management of long-gap esophageal atresia. Transl. Pediatr. 2024, 13, 329–342. [Google Scholar] [CrossRef]
- Borselle, D.; Davidson, J.; Loukogeorgakis, S.; De Coppi, P.; Patkowski, D. Thoracoscopic Stage Internal Traction Repair Reduces Time to Achieve Esophageal Continuity in Long Gap Esophageal Atresia. Eur. J. Pediatr. Surg. 2024, 34, 36–43. [Google Scholar] [CrossRef]
- Gallo, G.; Zwaveling, S.; Groen, H.; Van der Zee, D.; Hulscher, J. Long-gap esophageal atresia: A meta-analysis of jejunal interposition, colon interposition, and gastric pull-up. Eur. J. Pediatr. Surg. 2012, 22, 420–425. [Google Scholar] [CrossRef] [PubMed]
- van Hal, A.R.L.; Aanen, I.P.; Wijnen, R.M.H.; Pullens, B.; Vlot, J. The Value of Preoperative Rigid Tracheobronchoscopy for the Diagnosis of Tracheomalacia in Oesophageal Atresia Patients. J. Pediatr. Surg. 2024, 59, 161620. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, B. Endoscopy in Esophageal Atresia and Tracheoesophageal Fistula. Ann. Otol. Rhinol. Laryngol. 1981, 90, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Miyano, G.; Juang, D.; Sharp, N.E.; St Peter, S.D. Current patterns of practice and technique in the repair of esophageal atresia and tracheoesophageal fistua: An IPEG survey. J. Laparoendosc. Adv. Surg. Tech. A 2013, 23, 635–638. [Google Scholar] [CrossRef]
- Parolini, F.; Boroni, G.; Stefini, S.; Agapiti, C.; Bazzana, T.; Alberti, D. Role of preoperative tracheobronchoscopy in newborns with esophageal atresia: A review. World J. Gastrointest. Endosc. 2014, 6, 482–487. [Google Scholar] [CrossRef]
- Zani, A.; Eaton, S.; Hoellwarth, M.E.; Puri, P.; Tovar, J.; Fasching, G.; Bagolan, P.; Lukac, M.; Wijnen, R.; Kuebler, J.F.; et al. International survey on the management of esophageal atresia. Eur. J. Pediatr. Surg. 2014, 24, 3–8. [Google Scholar] [CrossRef]
- Mortellaro, V.E.; Fike, F.B.; Adibe, O.O.; Juang, D.; Aguayo, P.; Ostlie, D.J.; Holcomb, G.W.; St Peter, S.D. The use of high-frequency oscillating ventilation to facilitate stability during neonatal thoracoscopic operations. J. Laparoendosc. Adv. Surg. Tech. A 2011, 21, 877–879. [Google Scholar] [CrossRef]
- Bishay, M.; Giacomello, L.; Retrosi, G.; Thyoka, M.; Nah, S.A.; McHoney, M.; De Coppi, P.; Brierley, J.; Scuplak, S.; Kiely, E.M.; et al. Decreased cerebral oxygen saturation during thoracoscopic repair of congenital diaphragmatic hernia and esophageal atresia in infants. J. Pediatr. Surg. 2011, 46, 47–51. [Google Scholar] [CrossRef]
- Pierro, A. Hypercapnia and acidosis during the thoracoscopic repair of oesophageal atresia and congenital diaphragmatic hernia. J. Pediatr. Surg. 2015, 50, 247–249. [Google Scholar] [CrossRef]
- Bishay, M.; Giacomello, L.; Retrosi, G.; Thyoka, M.; Garriboli, M.; Brierley, J.; Harding, L.; Scuplak, S.; Cross, K.M.; Curry, J.I.; et al. Hypercapnia and acidosis during open and thoracoscopic repair of congenital diaphragmatic hernia and esophageal atresia: Results of a pilot randomized controlled trial. Ann. Surg. 2013, 258, 895–900. [Google Scholar] [CrossRef]
- Tytgat, S.H.; van Herwaarden, M.Y.; Stolwijk, L.J.; Keunen, K.; Benders, M.J.; de Graaff, J.C.; Milstein, D.M.; van der Zee, D.C.; Lemmers, P.M. Neonatal brain oxygenation during thoracoscopic correction of esophageal atresia. Surg. Endosc. 2016, 30, 2811–2817. [Google Scholar] [CrossRef] [PubMed]
- Zani, A.; Lamas-Pinheiro, R.; Paraboschi, I.; King, S.K.; Wolinska, J.; Zani-Ruttenstock, E.; Eaton, S.; Pierro, A. Intraoperative acidosis and hypercapnia during thoracoscopic repair of congenital diaphragmatic hernia and esophageal atresia/tracheoesophageal fistula. Paediatr. Anaesth. 2017, 27, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Sidler, M.; Wong, Z.H.; Eaton, S.; Ahmad, N.; Ong, M.; Morsi, A.; Rees, C.M.; Giuliani, S.; Blackburn, S.; Curry, J.I.; et al. Insufflation in minimally invasive surgery: Is there any advantage in staying low? J. Pediatr. Surg. 2020, 55, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Lin, Y.; He, Y.; Shen, Y.; Fan, J.; Fang, Y. Exploration of the thoracoscopic treatment of esophageal atresia under high-frequency ventilation. Front. Pediatr. 2022, 10, 1066492. [Google Scholar] [CrossRef]
- Spitz, L.; Kiely, E.; Brereton, R.J. Esophageal atresia: Five year experience with 148 cases. J. Pediatr. Surg. 1987, 22, 103–108. [Google Scholar] [CrossRef]
- O’Connell, J.S.; Janssen Lok, M.; Miyake, H.; Seo, S.; Bindi, E.; Alganabi, M.; Pierro, A. Post-operative paralysis and elective ventilation reduces anastomotic complications in esophageal atresia: A systematic review and meta-analysis. Pediatr. Surg. Int. 2019, 35, 87–95. [Google Scholar] [CrossRef]
- Davies, M.R.Q.; Beale, P.G. Protection of oesophageal anastomosis following uncomplicated repair of common-type oesophageal atresia by non-reversal of anaesthesia and graded withdrawal of respiratory support. Pediatr. Surg. Int. 1991, 6, 98–100. [Google Scholar] [CrossRef]
- Spitz, L.; Kiely, E.M.; Drake, D.P.; Pierro, A. Long-gap oesophageal atresia. Pediatr. Surg. Int. 1996, 11, 462–465. [Google Scholar] [CrossRef]
- Al-Salem, A.H.; Qaisaruddin, S.; Srair, H.A.; Dabbous, I.A.; Al-Hayek, R. Elective, postoperative ventilation in the management of esophageal atresia and tracheoesophageal fistula. Pediatr. Surg. Int. 1997, 12, 261–263. [Google Scholar] [CrossRef]
- Hseu, A.; Recko, T.; Jennings, R.; Nuss, R. Upper Airway Anomalies in Congenital Tracheoesophageal Fistula and Esophageal Atresia Patients. Ann. Otol. Rhinol. Laryngol. 2015, 124, 808–813. [Google Scholar] [CrossRef]
- Holcomb, G.W., 3rd; Rothenberg, S.S.; Bax, K.M.; Martinez-Ferro, M.; Albanese, C.T.; Ostlie, D.J.; van Der Zee, D.C.; Yeung, C.K. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula: A multi-institutional analysis. Ann. Surg. 2005, 242, 422–428; discussion 428–430. [Google Scholar] [CrossRef]
- Aworanti, O.M.; O’Connor, E.; Hannon, E.; Powis, M.; Alizai, N.; Crabbe, D.C.G. Extubation strategies after esophageal atresia repair. J. Pediatr. Surg. 2022, 57, 360–363. [Google Scholar] [CrossRef]
- Walor, D.; Berdon, W.; Anderson, N.; Holt, P.D.; Fox, M. Gaseous distention of the hypopharynx and cervical esophagus with nasal CPAP: A mimicker of pharyngeal perforation and esophageal atresia. Pediatr. Radiol. 2005, 35, 1196–1198. [Google Scholar] [CrossRef]
- Shah, P.S.; Gera, P.; Gollow, I.J.; Rao, S.C. Does continuous positive airway pressure for extubation in congenital tracheoesophageal fistula increase the risk of anastomotic leak? A retrospective cohort study. J. Paediatr. Child Health 2016, 52, 710–714. [Google Scholar] [CrossRef]
- Ferrand, A.; Roy, S.K.; Faure, C.; Moussa, A.; Aspirot, A. Postoperative noninvasive ventilation and complications in esophageal atresia-tracheoesophageal fistula. J. Pediatr. Surg. 2019, 54, 945–948. [Google Scholar] [CrossRef]
- De Rose, D.U.; Landolfo, F.; Giliberti, P.; Santisi, A.; Columbo, C.; Conforti, A.; Ronchetti, M.P.; Braguglia, A.; Dotta, A.; Capolupo, I.; et al. Post-operative ventilation strategies after surgical repair in neonates with esophageal atresia: A retrospective cohort study. J. Pediatr. Surg. 2022, 57, 801–805. [Google Scholar] [CrossRef]
- Williams, E.; Greenough, A. Respiratory Support of Infants with Congenital Diaphragmatic Hernia. Front. Pediatr. 2021, 9, 808317. [Google Scholar] [CrossRef]
- Zani, A.; Chung, W.K.; Deprest, J.; Harting, M.T.; Jancelewicz, T.; Kunisaki, S.M.; Patel, N.; Antounians, L.; Puligandla, P.S.; Keijzer, R. Congenital diaphragmatic hernia. Nat. Rev. Dis. Primers 2022, 8, 37. [Google Scholar] [CrossRef]
- Holden, K.I.; Ebanks, A.H.; Lally, K.P.; Harting, M.T. The CDH Study Group: Past, Present, and Future. Eur. J. Pediatr. Surg. 2024, 34, 162–171. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Gupta, V.S.; Lally, P.A.; Luco, M.; Tsao, K.; Lally, K.P.; Patel, N.; Harting, M.T. Early, Postnatal Pulmonary Hypertension Severity Predicts Inpatient Outcomes in Congenital Diaphragmatic Hernia. Neonatology 2021, 118, 147–154. [Google Scholar] [CrossRef]
- Chatterjee, D.; Ing, R.J.; Gien, J. Update on Congenital Diaphragmatic Hernia. Anesth. Analg. 2020, 131, 808–821. [Google Scholar] [CrossRef]
- Garne, E.; Haeusler, M.; Barisic, I.; Gjergja, R.; Stoll, C.; Clementi, M. Congenital diaphragmatic hernia: Evaluation of prenatal diagnosis in 20 European regions. Ultrasound. Obstet. Gynecol. 2002, 19, 329–333. [Google Scholar] [CrossRef]
- Mullassery, D.; Ba’ath, M.E.; Jesudason, E.C.; Losty, P.D. Value of liver herniation in prediction of outcome in fetal congenital diaphragmatic hernia: A systematic review and meta-analysis. Ultrasound. Obstet. Gynecol. 2010, 35, 609–614. [Google Scholar] [CrossRef]
- Jani, J.C.; Nicolaides, K.H.; Gratacós, E.; Vandecruys, H.; Deprest, J.A. Fetal lung-to-head ratio in the prediction of survival in severe left-sided diaphragmatic hernia treated by fetal endoscopic tracheal occlusion (FETO). Am. J. Obstet. Gynecol. 2006, 195, 1646–1650. [Google Scholar] [CrossRef]
- Deprest, J.A.; Flemmer, A.W.; Gratacos, E.; Nicolaides, K. Antenatal prediction of lung volume and in-utero treatment by fetal endoscopic tracheal occlusion in severe isolated congenital diaphragmatic hernia. Semin. Fetal. Neonatal. Med. 2009, 14, 8–13. [Google Scholar] [CrossRef]
- Victoria, T.; Bebbington, M.W.; Danzer, E.; Flake, A.W.; Johnson, M.P.; Dinan, D.; Adzick, N.S.; Hedrick, H.L. Use of magnetic resonance imaging in prenatal prognosis of the fetus with isolated left congenital diaphragmatic hernia. Prenat. Diagn. 2012, 32, 715–723. [Google Scholar] [CrossRef]
- Lee, T.C.; Lim, F.Y.; Keswani, S.G.; Frischer, J.S.; Haberman, B.; Kingma, P.S.; Habli, M.; Jaekle, R.K.; Sharp, G.; Kline-Fath, B.; et al. Late gestation fetal magnetic resonance imaging-derived total lung volume predicts postnatal survival and need for extracorporeal membrane oxygenation support in isolated congenital diaphragmatic hernia. J. Pediatr. Surg. 2011, 46, 1165–1171. [Google Scholar] [CrossRef]
- Harrison, M.R.; Keller, R.L.; Hawgood, S.B.; Kitterman, J.A.; Sandberg, P.L.; Farmer, D.L.; Lee, H.; Filly, R.A.; Farrell, J.A.; Albanese, C.T. A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N. Engl. J. Med. 2003, 349, 1916–1924. [Google Scholar] [CrossRef]
- Deprest, J.A.; Nicolaides, K.H.; Benachi, A.; Gratacos, E.; Ryan, G.; Persico, N.; Sago, H.; Johnson, A.; Wielgoś, M.; Berg, C.; et al. Randomized Trial of Fetal Surgery for Severe Left Diaphragmatic Hernia. N. Engl. J. Med. 2021, 385, 107–118. [Google Scholar] [CrossRef]
- Deprest, J.A.; Benachi, A.; Gratacos, E.; Nicolaides, K.H.; Berg, C.; Persico, N.; Belfort, M.; Gardener, G.J.; Ville, Y.; Johnson, A.; et al. Randomized Trial of Fetal Surgery for Moderate Left Diaphragmatic Hernia. N. Engl. J. Med. 2021, 385, 119–129. [Google Scholar] [CrossRef]
- Ali, K.; Bendapudi, P.; Polubothu, S.; Andradi, G.; Ofuya, M.; Peacock, J.; Hickey, A.; Davenport, M.; Nicolaides, K.; Greenough, A. Congenital diaphragmatic hernia-influence of fetoscopic tracheal occlusion on outcomes and predictors of survival. Eur. J. Pediatr. 2016, 175, 1071–1076. [Google Scholar] [CrossRef]
- McHugh, K.; Afaq, A.; Broderick, N.; Gabra, H.O.; Roebuck, D.J.; Elliott, M.J. Tracheomegaly: A complication of fetal endoscopic tracheal occlusion in the treatment of congenital diaphragmatic hernia. Pediatr. Radiol. 2010, 40, 674–680. [Google Scholar] [CrossRef]
- Breysem, L.; Debeer, A.; Claus, F.; Proesmans, M.; De Keyzer, F.; Lewi, P.; Allegaert, K.; Smet, M.H.; Deprest, J. Cross-sectional study of tracheomegaly in children after fetal tracheal occlusion for severe congenital diaphragmatic hernia. Radiology 2010, 257, 226–232. [Google Scholar] [CrossRef]
- Zani, A.; Sellars, M.; Allen, P.; Tyraskis, A.; Nicolaides, K.; Greenough, A.; Patel, S.; Davenport, M.; Ade-Ajayi, N. Tracheomegaly in infants with severe congenital diaphragmatic hernia treated with fetal endoluminal tracheal occlusion. J. Pediatr. 2014, 164, 1311–1315. [Google Scholar] [CrossRef]
- Basurto, D.; Watananirun, K.; Cordier, A.-G.; Otaño, J.; Carriere, D.; Scuglia, M.; de Luna Freire Vargas, A.M.; Prat, J.; Russo, F.M.; Debeer, A.; et al. Tracheomalacia and tracheomegaly in infants and children with congenital diaphragmatic hernia managed with and without fetoscopic endoluminal tracheal occlusion (FETO): A multicentre, retrospective cohort study. Lancet Child Adolesc. Health 2024, 8, 580–588. [Google Scholar] [CrossRef]
- Paut, O.; Mély, L.; Viard, L.; Silicani, M.A.; Guys, J.M.; Camboulives, J. Acute presentation of congenital diaphragmatic hernia past the neonatal period: A life threatening emergency. Can. J. Anaesth. 1996, 43, 621–625. [Google Scholar] [CrossRef]
- Cochius-den Otter, S.C.M.; Horn-Oudshoorn, E.J.J.; Allegaert, K.; DeKoninck, P.L.J.; Peters, N.C.J.; Cohen-Overbeek, T.E.; Reiss, I.K.M.; Tibboel, D. Routine Intubation in Newborns with Congenital Diaphragmatic Hernia. Pediatrics 2020, 146, e20201258. [Google Scholar] [CrossRef]
- Riley, J.S.; Antiel, R.M.; Rintoul, N.E.; Ades, A.M.; Waqar, L.N.; Lin, N.; Herkert, L.M.; D’Agostino, J.A.; Hoffman, C.; Peranteau, W.H.; et al. Reduced oxygen concentration for the resuscitation of infants with congenital diaphragmatic hernia. J. Perinatol. 2018, 38, 834–843. [Google Scholar] [CrossRef]
- Murthy, V.; D’Costa, W.; Nicolaides, K.; Davenport, M.; Fox, G.; Milner, A.D.; Campbell, M.; Greenough, A. Neuromuscular blockade and lung function during resuscitation of infants with congenital diaphragmatic hernia. Neonatology 2013, 103, 112–117. [Google Scholar] [CrossRef]
- Horn-Oudshoorn, E.J.J.; Knol, R.; Te Pas, A.B.; Hooper, S.B.; Cochius-den Otter, S.C.M.; Wijnen, R.M.H.; Schaible, T.; Reiss, I.K.M.; DeKoninck, P.L.J. Perinatal stabilisation of infants born with congenital diaphragmatic hernia: A review of current concepts. Arch. Dis. Child Fetal Neonatal Ed. 2020, 105, 449–454. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rakza, T.; Weslinck, N.; Vaast, P.; Houfflin-Debarge, V.; Mur, S.; Storme, L. Feasibility and safety of intact cord resuscitation in newborn infants with congenital diaphragmatic hernia (CDH). Resuscitation 2017, 120, 20–25. [Google Scholar] [CrossRef]
- Foglia, E.E.; Ades, A.; Hedrick, H.L.; Rintoul, N.; Munson, D.A.; Moldenhauer, J.; Gebb, J.; Serletti, B.; Chaudhary, A.; Weinberg, D.D.; et al. Initiating resuscitation before umbilical cord clamping in infants with congenital diaphragmatic hernia: A pilot feasibility trial. Arch. Dis. Child Fetal Neonatal Ed. 2020, 105, 322–326. [Google Scholar] [CrossRef]
- Horn-Oudshoorn, E.J.J.; Vermeulen, M.J.; Knol, R.; Bout-Rebel, R.; Te Pas, A.B.; Hooper, S.B.; Otter, S.; Wijnen, R.M.H.; Crossley, K.J.; Rafat, N.; et al. Multicentre, randomised controlled trial of physiological-based cord clamping versus immediate cord clamping in infants with a congenital diaphragmatic hernia (PinC): Statistical analysis plan. Trials 2024, 25, 198. [Google Scholar] [CrossRef]
- Congenital Diaphragmatic Hernia Study Group. Estimating disease severity of congenital diaphragmatic hernia in the first 5 minutes of life. J. Pediatr. Surg. 2001, 36, 141–145. [Google Scholar] [CrossRef]
- Schultz, C.M.; DiGeronimo, R.J.; Yoder, B.A. Congenital diaphragmatic hernia: A simplified postnatal predictor of outcome. J. Pediatr. Surg. 2007, 42, 510–516. [Google Scholar] [CrossRef]
- Sinha, C.K.; Islam, S.; Patel, S.; Nicolaides, K.; Greenough, A.; Davenport, M. Congenital diaphragmatic hernia: Prognostic indices in the fetal endoluminal tracheal occlusion era. J. Pediatr. Surg. 2009, 44, 312–316. [Google Scholar] [CrossRef]
- Oh, C.; Youn, J.K.; Han, J.W.; Yang, H.B.; Lee, S.; Seo, J.M.; Ho, I.G.; Kim, S.H.; Cho, Y.H.; Shin, S.H.; et al. Predicting Survival of Congenital Diaphragmatic Hernia on the First Day of Life. World J. Surg. 2019, 43, 282–290. [Google Scholar] [CrossRef]
- Ruttenstock, E.; Wright, N.; Barrena, S.; Krickhahn, A.; Castellani, C.; Desai, A.P.; Rintala, R.; Tovar, J.; Till, H.; Zani, A.; et al. Best oxygenation index on day 1: A reliable marker for outcome and survival in infants with congenital diaphragmatic hernia. Eur. J. Pediatr. Surg. 2015, 25, 3–8. [Google Scholar] [CrossRef]
- Tan, Y.W.; Adamson, L.; Forster, C.; Davies, B.; Sharkey, D. Using serial oxygenation index as an objective predictor of survival for antenatally diagnosed congenital diaphragmatic hernia. J. Pediatr. Surg. 2012, 47, 1984–1989. [Google Scholar] [CrossRef]
- Tan, Y.W.; Ali, K.; Andradi, G.; Sasidharan, L.; Greenough, A.; Davenport, M. Prognostic value of the oxygenation index to predict survival and timing of surgery in infants with congenital diaphragmatic hernia. J. Pediatr. Surg. 2019, 54, 1567–1572. [Google Scholar] [CrossRef]
- Wung, J.T.; James, L.S.; Kilchevsky, E.; James, E. Management of infants with severe respiratory failure and persistence of the fetal circulation, without hyperventilation. Pediatrics 1985, 76, 488–494. [Google Scholar] [CrossRef]
- Logan, J.W.; Cotten, C.M.; Goldberg, R.N.; Clark, R.H. Mechanical ventilation strategies in the management of congenital diaphragmatic hernia. Semin. Pediatr. Surg. 2007, 16, 115–125. [Google Scholar] [CrossRef]
- Farkouh-Karoleski, C.; Najaf, T.; Wynn, J.; Aspelund, G.; Chung, W.K.; Stolar, C.J.; Mychaliska, G.B.; Warner, B.W.; Wagner, A.J.; Cusick, R.A.; et al. A definition of gentle ventilation in congenital diaphragmatic hernia: A survey of neonatologists and pediatric surgeons. J. Perinat. Med. 2017, 45, 1031–1038. [Google Scholar] [CrossRef]
- Guidry, C.A.; Hranjec, T.; Rodgers, B.M.; Kane, B.; McGahren, E.D. Permissive hypercapnia in the management of congenital diaphragmatic hernia: Our institutional experience. J. Am. Coll. Surg. 2012, 214, 640–647.e1. [Google Scholar] [CrossRef]
- Snoek, K.G.; Reiss, I.K.; Greenough, A.; Capolupo, I.; Urlesberger, B.; Wessel, L.; Storme, L.; Deprest, J.; Schaible, T.; van Heijst, A.; et al. Standardized Postnatal Management of Infants with Congenital Diaphragmatic Hernia in Europe: The CDH EURO Consortium Consensus—2015 Update. Neonatology 2016, 110, 66–74. [Google Scholar] [CrossRef]
- Snoek, K.G.; Capolupo, I.; van Rosmalen, J.; Hout Lde, J.; Vijfhuize, S.; Greenough, A.; Wijnen, R.M.; Tibboel, D.; Reiss, I.K. Conventional Mechanical Ventilation Versus High-frequency Oscillatory Ventilation for Congenital Diaphragmatic Hernia: A Randomized Clinical Trial (The VICI-trial). Ann. Surg. 2016, 263, 867–874. [Google Scholar] [CrossRef]
- Lee, R.; Hunt, K.A.; Williams, E.E.; Dassios, T.; Greenough, A. Work of breathing at different tidal volume targets in newborn infants with congenital diaphragmatic hernia. Eur. J. Pediatr. 2022, 181, 2453–2458. [Google Scholar] [CrossRef]
- Puligandla, P.S.; Grabowski, J.; Austin, M.; Hedrick, H.; Renaud, E.; Arnold, M.; Williams, R.F.; Graziano, K.; Dasgupta, R.; McKee, M.; et al. Management of congenital diaphragmatic hernia: A systematic review from the APSA outcomes and evidence based practice committee. J. Pediatr. Surg. 2015, 50, 1958–1970. [Google Scholar] [CrossRef]
- Van Meurs, K. Is surfactant therapy beneficial in the treatment of the term newborn infant with congenital diaphragmatic hernia? J. Pediatr. 2004, 145, 312–316. [Google Scholar] [CrossRef]
- Lally, K.P.; Lally, P.A.; Langham, M.R.; Hirschl, R.; Moya, F.R.; Tibboel, D.; Van Meurs, K. Surfactant does not improve survival rate in preterm infants with congenital diaphragmatic hernia. J. Pediatr. Surg. 2004, 39, 829–833. [Google Scholar] [CrossRef]
- Colby, C.E.; Lally, K.P.; Hintz, S.R.; Lally, P.A.; Tibboel, D.; Moya, F.R.; VanMeurs, K.P. Surfactant replacement therapy on ECMO does not improve outcome in neonates with congenital diaphragmatic hernia. J. Pediatr. Surg. 2004, 39, 1632–1637. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.C.; Boutin, M.A.; Knodel, E.M.; Proudfoot, J.A.; Lane, B.P.; Evans, M.L.; Suttner, D.M.; Kimball, A.L. Heliox Adjunct Therapy for Neonates with Congenital Diaphragmatic Hernia. Respir. Care 2018, 63, 1147–1153. [Google Scholar] [CrossRef]
- Hirschl, R.B.; Philip, W.F.; Glick, L.; Greenspan, J.; Smith, K.; Thompson, A.; Wilson, J.; Adzick, N.S. A prospective, randomized pilot trial of perfluorocarbon-induced lung growth in newborns with congenital diaphragmatic hernia. J. Pediatr. Surg. 2003, 38, 283–289; discussion 283–289. [Google Scholar] [CrossRef] [PubMed]
- Greenough, A. Management of infants with congenital diaphragmatic hernia and pulmonary hypertension-one size does not fit all. Pediatr. Res. 2023, 93, 1795–1796. [Google Scholar] [CrossRef] [PubMed]
- Putnam, L.R.; Tsao, K.; Morini, F.; Lally, P.A.; Miller, C.C.; Lally, K.P.; Harting, M.T. Evaluation of Variability in Inhaled Nitric Oxide Use and Pulmonary Hypertension in Patients with Congenital Diaphragmatic Hernia. JAMA Pediatr. 2016, 170, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Surak, A.; Mahgoub, L.; Ting, J.Y. Hemodynamic management of congenital diaphragmatic hernia: The role of targeted neonatal echocardiography. World J. Pediatr. Surg. 2024, 7, e000790. [Google Scholar] [CrossRef]
- The Neonatal Inhaled Nitric Oxide Study Group (NINOS). Inhaled nitric oxide and hypoxic respiratory failure in infants with congenital diaphragmatic hernia. Pediatrics 1997, 99, 838–845. [Google Scholar] [CrossRef]
- Noh, C.Y.; Chock, V.Y.; Bhombal, S.; Danzer, E.; Patel, N.; Dahlen, A.; Harting, M.T.; Lally, K.P.; Ebanks, A.H.; Van Meurs, K.P. Early nitric oxide is not associated with improved outcomes in congenital diaphragmatic hernia. Pediatr. Res. 2023, 93, 1899–1906. [Google Scholar] [CrossRef]
- Lawrence, K.M.; Monos, S.; Adams, S.; Herkert, L.; Peranteau, W.H.; Munson, D.A.; Hopper, R.K.; Avitabile, C.M.; Rintoul, N.E.; Hedrick, H.L. Inhaled Nitric Oxide Is Associated with Improved Oxygenation in a Subpopulation of Infants with Congenital Diaphragmatic Hernia and Pulmonary Hypertension. J. Pediatr. 2020, 219, 167–172. [Google Scholar] [CrossRef]
- Thodika, F.; Dimitrova, S.; Nanjundappa, M.; Davenport, M.; Nicolaides, K.; Dassios, T.; Greenough, A. Prediction of survival in infants with congenital diaphragmatic hernia and the response to inhaled nitric oxide. Eur. J. Pediatr. 2022, 181, 3683–3689. [Google Scholar] [CrossRef]
- Cochius-den Otter, S.; Schaible, T.; Greenough, A.; van Heijst, A.; Patel, N.; Allegaert, K.; van Rosmalen, J.; Tibboel, D. The CoDiNOS trial protocol: An international randomised controlled trial of intravenous sildenafil versus inhaled nitric oxide for the treatment of pulmonary hypertension in neonates with congenital diaphragmatic hernia. BMJ Open 2019, 9, e032122. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Keszler, M.; Kirpalani, H.; Van Meurs, K.; Chess, P.; Ambalavanan, N.; Yoder, B.; Fraga, M.V.; Hedrick, H.; Lally, K.P.; et al. Milrinone in congenital diaphragmatic hernia—A randomized pilot trial: Study protocol, review of literature and survey of current practices. Matern. Health Neonatol. Perinatol. 2017, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Breaux, C.W., Jr.; Rouse, T.M.; Cain, W.S.; Georgeson, K.E. Improvement in survival of patients with congenital diaphragmatic hernia utilizing a strategy of delayed repair after medical and/or extracorporeal membrane oxygenation stabilization. J. Pediatr. Surg. 1991, 26, 333–336; discussion 336–338. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Bösenberg, A.T. Evolving management of congenital diaphragmatic hernia. Paediatr. Anaesth. 2007, 17, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Fennessy, P.; Crowe, S.; Lenihan, M.; Healy, M. Anesthesia consensus on clinical parameters for the timing of surgical repair in congenital diaphragmatic hernia. Paediatr. Anaesth. 2018, 28, 751–752. [Google Scholar] [CrossRef]
- Deeney, S.; Howley, L.W.; Hodges, M.; Liechty, K.W.; Marwan, A.I.; Gien, J.; Kinsella, J.P.; Crombleholme, T.M. Impact of Objective Echocardiographic Criteria for Timing of Congenital Diaphragmatic Hernia Repair. J. Pediatr. 2018, 192, 99–104.e104. [Google Scholar] [CrossRef]
- Robertson, J.O.; Criss, C.N.; Hsieh, L.B.; Matsuko, N.; Gish, J.S.; Mon, R.A.; Johnson, K.N.; Hirschl, R.B.; Mychaliska, G.B.; Gadepalli, S.K. Comparison of early versus delayed strategies for repair of congenital diaphragmatic hernia on extracorporeal membrane oxygenation. J. Pediatr. Surg. 2018, 53, 629–634. [Google Scholar] [CrossRef]
- Delaplain, P.T.; Harting, M.T.; Jancelewicz, T.; Zhang, L.; Yu, P.T.; Di Nardo, M.; Chen, Y.; Stein, J.E.; Ford, H.R.; Nguyen, D.V.; et al. Potential survival benefit with repair of congenital diaphragmatic hernia (CDH) after extracorporeal membrane oxygenation (ECMO) in select patients: Study by ELSO CDH Interest Group. J. Pediatr. Surg. 2019, 54, 1132–1137. [Google Scholar] [CrossRef]
- Strumiłło, B.; Jóźwiak, A.; Maroszyńska, I.; Piaseczna-Piotrowska, A. Repair of congenital diaphragmatic hernia on extracorporeal membrane oxygenation-observations of a paediatric surgeon. Kardiochir. Torakochirurgia Pol. 2022, 19, 16–21. [Google Scholar] [CrossRef]
- Schmoke, N.; Rose, A.; Nemeh, C.; Wu, Y.S.; Wang, P.; Kurlansky, P.; Neunert, C.; Middlesworth, W.; Duron, V. Optimizing Congenital Diaphragmatic Hernia Repair on ECMO: Evaluating the Risk of Bleeding. J. Pediatr. Surg. 2024, 59, 161766. [Google Scholar] [CrossRef]
- Okazaki, T.; Okawada, M.; Ishii, J.; Koga, H.; Miyano, G.; Doi, T.; Ogasawara, Y.; Lane, G.J.; Yamataka, A. Intraoperative ventilation during thoracoscopic repair of neonatal congenital diaphragmatic hernia. Pediatr. Surg. Int. 2017, 33, 1097–1101. [Google Scholar] [CrossRef]
- Zamakhshary, M.; Mah, K.; Mah, D.; Cameron, B.; Bohn, D.; Bass, J.; Scott, L.; Kim, P.C. Physiologic predictors for the need for patch closure in neonatal congenital diaphragmatic hernia. Pediatr. Surg. Int. 2008, 24, 667–670. [Google Scholar] [CrossRef]
- Moss, R.L.; Chen, C.M.; Harrison, M.R. Prosthetic patch durability in congenital diaphragmatic hernia: A long-term follow-up study. J. Pediatr. Surg. 2001, 36, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Brindle, M.E.; Brar, M.; Skarsgard, E.D. Patch repair is an independent predictor of morbidity and mortality in congenital diaphragmatic hernia. Pediatr. Surg. Int. 2011, 27, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Vijfhuize, S.; Deden, A.C.; Costerus, S.A.; Sloots, C.E.; Wijnen, R.M. Minimal access surgery for repair of congenital diaphragmatic hernia: Is it advantageous?--An open review. Eur. J. Pediatr. Surg. 2012, 22, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Terui, K.; Nagata, K.; Ito, M.; Yamoto, M.; Shiraishi, M.; Taguchi, T.; Hayakawa, M.; Okuyama, H.; Yoshida, H.; Masumoto, K.; et al. Surgical approaches for neonatal congenital diaphragmatic hernia: A systematic review and meta-analysis. Pediatr. Surg. Int. 2015, 31, 891–897. [Google Scholar] [CrossRef]
- Poole, G.; Shetty, S.; Greenough, A. The use of neurally-adjusted ventilatory assist (NAVA) for infants with congenital diaphragmatic hernia (CDH). J. Perinat. Med. 2022, 50, 1163–1167. [Google Scholar] [CrossRef]
- Kurland, Y.; Gurung, K.; Pallotto, E.K.; Manimtim, W.; Feldman, K.; Staggs, V.S.; Truog, W. Neurally adjusted ventilatory assist in neonates with congenital diaphragmatic hernia. J. Perinatol. 2021, 41, 1910–1915. [Google Scholar] [CrossRef]
- Oda, A.; Lehtonen, L.; Soukka, H. Neurally adjusted ventilatory assist can be used to wean infants with congenital diaphragmatic hernias off respiratory support. Acta Paediatr. 2018, 107, 718–719. [Google Scholar] [CrossRef]
- Amin, R.; Arca, M.J. Feasibility of Non-invasive Neurally Adjusted Ventilator Assist After Congenital Diaphragmatic Hernia Repair. J. Pediatr. Surg. 2019, 54, 434–438. [Google Scholar] [CrossRef]
- Meinen, R.D.; Alali, Y.I.; Al-Subu, A.; Wilhelm, M.; Wraight, C.L.; McAdams, R.M.; Limjoco, J.J.; McCulley, D.J. Neurally-Adjusted Ventilatory Assist Can Facilitate Extubation in Neonates with Congenital Diaphragmatic Hernia. Respir. Care 2021, 66, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Weems, M.F.; Grover, T.R.; Seabrook, R.; DiGeronimo, R.; Gien, J.; Keene, S.; Rintoul, N.; Daniel, J.M.; Johnson, Y.; Guner, Y.; et al. Analgesia, Sedation, and Neuromuscular Blockade in Infants with Congenital Diaphragmatic Hernia. Am. J. Perinatol. 2023, 40, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Pederiva, F.; Rothenberg, S.S.; Hall, N.; Ijsselstijn, H.; Wong, K.K.Y.; von der Thüsen, J.; Ciet, P.; Achiron, R.; Pio d’Adamo, A.; Schnater, J.M. Congenital lung malformations. Nat. Rev. Dis. Primers 2023, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.T.; Kan, A.; Shek, N.; Tam, P.; Wong, K.K. Is congenital pulmonary airway malformation really a rare disease? Result of a prospective registry with universal antenatal screening program. Pediatr. Surg. Int. 2017, 33, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Chaudhary, K.; Hayaran, N.; Neogi, S. Anesthetic considerations in patients with cystic pulmonary adenomatoid malformations. J. Anaesthesiol. Clin. Pharmacol. 2021, 37, 146–152. [Google Scholar] [CrossRef]
- Chiluveru, S.A.; Dave, N.M.; Dias, R.J.; Garasia, M.B. Congenital pulmonary airway malformation with atrial septal defect and pulmonary hypertension for lobectomy-anesthetic considerations. Ann. Card. Anaesth. 2016, 19, 372–374. [Google Scholar] [CrossRef]
- Adzick, N.S.; Harrison, M.R.; Glick, P.L.; Golbus, M.S.; Anderson, R.L.; Mahony, B.S.; Callen, P.W.; Hirsch, J.H.; Luthy, D.A.; Filly, R.A.; et al. Fetal cystic adenomatoid malformation: Prenatal diagnosis and natural history. J. Pediatr. Surg. 1985, 20, 483–488. [Google Scholar] [CrossRef]
- Kunisaki, S.M.; Saito, J.M.; Fallat, M.E.; Peter, S.D.S.; Lal, D.R.; Karmakar, M.; Deans, K.J.; Gadepalli, S.K.; Hirschl, R.B.; Minneci, P.C.; et al. Fetal Risk Stratification and Outcomes in Children with Prenatally Diagnosed Lung Malformations: Results from a Multi-Institutional Research Collaborative. Ann. Surg. 2022, 276, e622–e630. [Google Scholar] [CrossRef]
- Crombleholme, T.M.; Coleman, B.; Hedrick, H.; Liechty, K.; Howell, L.; Flake, A.W.; Johnson, M.; Adzick, N.S. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J. Pediatr. Surg. 2002, 37, 331–338. [Google Scholar] [CrossRef]
- Kane, S.C.; Ancona, E.; Reidy, K.L.; Palma-Dias, R. The Utility of the Congenital Pulmonary Airway Malformation-Volume Ratio in the Assessment of Fetal Echogenic Lung Lesions: A Systematic Review. Fetal. Diagn. Ther. 2020, 47, 171–181. [Google Scholar] [CrossRef]
- Gerall, C.; Chumdermpadestuk, R.; Jacobs, S.; Weijia, F.; Maddocks, A.; Ayyala, R.; Miller, R.; Simpson, L.; Rothenberg, S.; Duron, V. Prenatal ultrasound-and MRI-based imaging predictors of respiratory symptoms at birth for congenital lung malformations. J. Pediatr. Surg. 2023, 58, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Delacourt, C.; Bertille, N.; Salomon, L.J.; Rahshenas, M.; Benachi, A.; Bonnard, A.; Choupeaux, L.; Fouquet, V.; Goua, V.; Hameury, F.; et al. Predicting the risk of respiratory distress in newborns with congenital pulmonary malformations. Eur. Respir. J. 2022, 59, 2100949. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.C.J.; Hijkoop, A.; Hermelijn, S.M.; van Schoonhoven, M.M.; Eggink, A.J.; van Rosmalen, J.; Otter, S.; Tibboel, D.; IJsselstijn, H.; Schnater, J.M.; et al. Prediction of postnatal outcome in fetuses with congenital lung malformation: 2-year follow-up study. Ultrasound Obstet. Gynecol. 2021, 58, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Ruchonnet-Metrailler, I.; Leroy-Terquem, E.; Stirnemann, J.; Cros, P.; Ducoin, H.; Hadchouel, A.; Khen-Dunlop, N.; Labbé, A.; Labouret, G.; Lebras, M.N.; et al. Neonatal outcomes of prenatally diagnosed congenital pulmonary malformations. Pediatrics 2014, 133, e1285–e1291. [Google Scholar] [CrossRef]
- Montgomery, A.; Peiffer, S.; Mehl, S.; Lee, T.C.; Keswani, S.G.; King, A. Management and Outcomes of Patients with High-Risk (Congenital Lung Malformation Volume Ratio ≥ 1.6) Congenital Lung Malformations. J. Surg. Res. 2024, 295, 559–566. [Google Scholar] [CrossRef]
- Mehl, S.C.; Short, W.D.; Kinley, A.; Olutoye, O.O., 2nd; Lee, T.C.; Keswani, S.G.; King, A. Maternal Steroids in High-Risk Congenital Lung Malformations. J. Surg. Res. 2022, 280, 312–319. [Google Scholar] [CrossRef]
- Morris, L.M.; Lim, F.Y.; Livingston, J.C.; Polzin, W.J.; Crombleholme, T.M. High-risk fetal congenital pulmonary airway malformations have a variable response to steroids. J. Pediatr. Surg. 2009, 44, 60–65. [Google Scholar] [CrossRef]
- Peranteau, W.H.; Wilson, R.D.; Liechty, K.W.; Johnson, M.P.; Bebbington, M.W.; Hedrick, H.L.; Flake, A.W.; Adzick, N.S. Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn. Ther. 2007, 22, 365–371. [Google Scholar] [CrossRef]
- Tsao, K.; Hawgood, S.; Vu, L.; Hirose, S.; Sydorak, R.; Albanese, C.T.; Farmer, D.L.; Harrison, M.R.; Lee, H. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J. Pediatr. Surg. 2003, 38, 508–510. [Google Scholar] [CrossRef]
- Gallagher, L.T.; Lyttle, B.D.; Dawson-Gore, C.; Vaughn, A.E.; Breckenfelder, C.; Reynolds, R.; Zaretsky, M.V.; Derderian, S.C. The Effect of Steroids on Prenatally Diagnosed Lung Lesions. J. Pediatr. Surg. 2024, 59, 969–974. [Google Scholar] [CrossRef]
- Peranteau, W.H.; Adzick, N.S.; Boelig, M.M.; Flake, A.W.; Hedrick, H.L.; Howell, L.J.; Moldenhauer, J.S.; Khalek, N.; Martinez-Poyer, J.; Johnson, M.P. Thoracoamniotic shunts for the management of fetal lung lesions and pleural effusions: A single-institution review and predictors of survival in 75 cases. J. Pediatr. Surg. 2015, 50, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, O.; Furman, Y.; Kimhi, G.; Leibovitch, L.; Mazkereth, R.; Yinon, Y.; Lipitz, S.; Strauss, T.; Weisz, B. In-utero treatment of prenatal thoracic abnormalities by thoraco-amniotic shunts, short and long term neuro developmental outcome: A single center experience. J. Pediatr. Surg. 2022, 57, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.; Cazacu, R.; Ade-Ajayi, N.; Patel, S.B.; Nicolaides, K.; Davenport, M. The long-term outcome following thoraco-amniotic shunting for congenital lung malformations. J. Pediatr. Surg. 2023, 58, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Mallmann, M.R.; Geipel, A.; Bludau, M.; Matil, K.; Gottschalk, I.; Hoopmann, M.; Müller, A.; Bachour, H.; Heydweiller, A.; Gembruch, U.; et al. Bronchopulmonary sequestration with massive pleural effusion: Pleuroamniotic shunting vs intrafetal vascular laser ablation. Ultrasound Obstet. Gynecol. 2014, 44, 441–446. [Google Scholar] [CrossRef]
- Laberge, J.M.; Puligandla, P.; Flageole, H. Asymptomatic congenital lung malformations. Semin. Pediatr. Surg. 2005, 14, 16–33. [Google Scholar] [CrossRef]
- Muntean, A.; Banias, L.E.; Ade-Ajayi, N.; Patel, S.B.; McKinney, O.; Davenport, M. Neonatal congenital pulmonary airway malformation associated with mucinous adenocarcinoma and KRAS mutations. J. Pediatr. Surg. 2022, 57, 520–526. [Google Scholar] [CrossRef]
- Guruswamy, V.; Roberts, S.; Arnold, P.; Potter, F. Anaesthetic management of a neonate with congenital cyst adenoid malformation. Br. J. Anaesth. 2005, 95, 240–242. [Google Scholar] [CrossRef]
- Takrouri, M.S.; Maghaireh, A.; Obeidat, M.R. Failed sevoflurane induction of anesthesia in a child affected by congenital cystic adenomatoid malformation of the lungs. Anesth. Essays Res. 2010, 4, 44–45. [Google Scholar] [CrossRef]
- Lee, J.H.; Bae, J.I.; Jang, Y.E.; Kim, E.H.; Kim, H.S.; Kim, J.T. Lung protective ventilation during pulmonary resection in children: A prospective, single-centre, randomised controlled trial. Br. J. Anaesth. 2019, 122, 692–701. [Google Scholar] [CrossRef]
- Templeton, T.W.; Piccioni, F.; Chatterjee, D. An Update on One-Lung Ventilation in Children. Anesth. Analg. 2021, 132, 1389–1399. [Google Scholar] [CrossRef]
- Purohit, A.; Bhargava, S.; Mangal, V.; Parashar, V.K. Lung isolation, one-lung ventilation and hypoxaemia during lung isolation. Indian J. Anaesth. 2015, 59, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, H.; Hatori, E.; Takeda, J.; Morisaki, H. Anesthetic management of a neonate with congenital cystic adenomatoid malformation. Masui 2014, 63, 101–104. [Google Scholar] [PubMed]
- Mei-Zahav, M.; Konen, O.; Manson, D.; Langer, J.C. Is congenital lobar emphysema a surgical disease? J. Pediatr. Surg. 2006, 41, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Davenport, M. The argument for operative approach to asymptomatic lung lesions. Semin. Pediatr. Surg. 2015, 24, 187–195. [Google Scholar] [CrossRef]
- Stanton, M. The argument for a non-operative approach to asymptomatic lung lesions. Semin. Pediatr. Surg. 2015, 24, 183–186. [Google Scholar] [CrossRef]
- Tsai, A.Y.; Liechty, K.W.; Hedrick, H.L.; Bebbington, M.; Wilson, R.D.; Johnson, M.P.; Howell, L.J.; Flake, A.W.; Adzick, N.S. Outcomes after postnatal resection of prenatally diagnosed asymptomatic cystic lung lesions. J. Pediatr. Surg. 2008, 43, 513–517. [Google Scholar] [CrossRef]
- Rothenberg, S.S.; Middlesworth, W.; Kadennhe-Chiweshe, A.; Aspelund, G.; Kuenzler, K.; Cowles, R.; Bodenstein, L.; Kay, S.; Shipman, K.; Rothenberg, C.; et al. Two decades of experience with thoracoscopic lobectomy in infants and children: Standardizing techniques for advanced thoracoscopic surgery. J. Laparoendosc. Adv. Surg. Tech. A 2015, 25, 423–428. [Google Scholar] [CrossRef]
- Kapralik, J.; Wayne, C.; Chan, E.; Nasr, A. Surgical versus conservative management of congenital pulmonary airway malformation in children: A systematic review and meta-analysis. J. Pediatr. Surg. 2016, 51, 508–512. [Google Scholar] [CrossRef]
- Berrington de Gonzalez, A.; Pasqual, E.; Veiga, L. Epidemiological studies of CT scans and cancer risk: The state of the science. Br. J. Radiol. 2021, 94, 20210471. [Google Scholar] [CrossRef]
- Kersten, C.M.; Hermelijn, S.M.; Dossche, L.W.J.; Muthialu, N.; Losty, P.D.; Schurink, M.; Rietman, A.B.; Poley, M.J.; van Rosmalen, J.; Zanen-van den Adel, T.P.L.; et al. COllaborative Neonatal Network for the first European CPAM Trial (CONNECT): A study protocol for a randomised controlled trial. BMJ Open 2023, 13, e071989. [Google Scholar] [CrossRef]
- Victoria, T.; Andronikou, S.; Bowen, D.; Laje, P.; Weiss, D.A.; Johnson, A.M.; Peranteau, W.H.; Canning, D.A.; Adzick, N.S. Fetal anterior abdominal wall defects: Prenatal imaging by magnetic resonance imaging. Pediatr. Radiol. 2018, 48, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Fogelström, A.; Caldeman, C.; Oddsberg, J.; Löf Granström, A.; Mesas Burgos, C. Omphalocele: National current birth prevalence and survival. Pediatr. Surg. Int. 2021, 37, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Kilby, M.D. The incidence of gastroschisis. BMJ 2006, 332, 250–251. [Google Scholar] [CrossRef] [PubMed]
- Panitch, H.B. Pulmonary complications of abdominal wall defects. Paediatr. Respir. Rev. 2015, 16, 11–17. [Google Scholar] [CrossRef]
- Langer, J.C. Chapter 79: Omphalocele. In Pediatric Surgery: General Principles and Newborn Surgery; Puri, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Schwart, M.Z.; Timmapuri, S.J. Chapter 80: Gastroschisis. In Pediatric Surgery: General Principles and Newborn Surgery; Puri, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Arlen, A.M.; Nawaf, C.; Kirsch, A.J. Prune belly syndrome: Current perspectives. Pediatr. Health Med. Ther. 2019, 10, 75–81. [Google Scholar] [CrossRef]
- Apostel, H.; Duval, E.; De Dooy, J.; Jorens, P.G.; Schepens, T. Respiratory support in the absence of abdominal muscles: A case study of ventilatory management in prune belly syndrome. Paediatr. Respir. Rev. 2021, 37, 44–47. [Google Scholar] [CrossRef]
- De Troyer, A.; Sampson, M.; Sigrist, S.; Macklem, P.T. Action of costal and crural parts of the diaphragm on the rib cage in dog. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1982, 53, 30–39. [Google Scholar] [CrossRef]
- Hershenson, M.B.; Brouillette, R.T.; Klemka, L.; Raffensperger, J.D.; Poznanski, A.K.; Hunt, C.E. Respiratory insufficiency in newborns with abdominal wall defects. J. Pediatr. Surg. 1985, 20, 348–353. [Google Scholar] [CrossRef]
- Griscom, N.; Driscoll, S. Radiography of stillborn fetuses and infants dying at birth. Am. J. Roentgenol. 1980, 134, 485–489. [Google Scholar] [CrossRef]
- Argyle, J.C. Pulmonary hypoplasia in infants with giant abdominal wall defects. Pediatr. Pathol. 1989, 9, 43–55. [Google Scholar] [CrossRef]
- Ein, S.H.; Langer, J.C. Delayed management of giant omphalocele using silver sulfadiazine cream: An 18-year experience. J. Pediatr. Surg. 2012, 47, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.S.; Stamilio, D.M.; Dicke, J.M.; Gray, D.L.; Macones, G.A.; Odibo, A.O. Predicting adverse neonatal outcomes in fetuses with abdominal wall defects using prenatal risk factors. Am. J. Obstet. Gynecol. 2009, 201, 383.e1–383.e6. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, M.; Rafferty, G.F.; Davenport, M.; Persico, N.; Jani, J.; Nicolaides, K.; Greenough, A. Three-dimensional ultrasound fetal lung volumes and infant respiratory outcome: A prospective observational study. Bjog 2011, 118, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.; Dassios, T.; Bothamley, Z.; Haque, S.; Watson, C.; Davenport, M.; Harris, C.; Greenough, A. Prediction of bronchopulmonary dysplasia by the chest radiographic thoracic area on day one in infants with exomphalos. J. Perinat. Med. 2024, 52, 429–432. [Google Scholar] [CrossRef]
- Thompson, P.J.; Greenough, A.; Dykes, E.; Nicolaides, K.H. Impaired respiratory function in infants with anterior abdominal wall defects. J. Pediatr. Surg. 1993, 28, 664–666. [Google Scholar] [CrossRef]
- Di Marco, F.; Rota Sperti, L.; Milan, B.; Stucchi, R.; Centanni, S.; Brochard, L.; Fumagalli, R. Measurement of functional residual capacity by helium dilution during partial support ventilation: In vitro accuracy and in vivo precision of the method. Intensive Care Med. 2007, 33, 2109–2115. [Google Scholar] [CrossRef]
- Laubscher, B.; Greenough, A.; Dimitriou, G.; Davenport, M.; Nicolaides, K.H. Serial lung volume measurements during the perinatal period in infants with abdominal wall defects. J. Pediatr. Surg. 1998, 33, 497–499. [Google Scholar] [CrossRef]
- Jenkinson, A.; Krishnan, M.; Davenport, M.; Harris, C.; Dassios, T.; Greenough, A. Chest radiographic thoracic areas and respiratory outcomes in infants with anterior abdominal wall defects. J. Perinat. Med. 2024, 52, 552–555. [Google Scholar] [CrossRef]
- Dimitriou, G.; Greenough, A.; Giffin, F.; Davenport, M.; Nicolaides, K.H. Temporary impairment of lung function in infants with anterior abdominal wall defects who have undergone surgery. J. Pediatr. Surg. 1996, 31, 670–672. [Google Scholar] [CrossRef]
- Towne, B.H.; Peters, G.; Chang, J.H.T. The problem of “giant” omphalocele. J. Pediatr. Surg. 1980, 15, 543–548. [Google Scholar] [CrossRef]
- Kamata, S.; Usui, N.; Sawai, T.; Nose, K.; Fukuzawa, M. Prenatal detection of pulmonary hypoplasia in giant omphalocele. Pediatr. Surg. Int. 2008, 24, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Danzer, E.; Victoria, T.; Bebbington, M.W.; Siegle, J.; Rintoul, N.E.; Johnson, M.P.; Flake, A.W.; Adzick, N.S.; Hedrick, H.L. Fetal MRI-calculated total lung volumes in the prediction of short-term outcome in giant omphalocele: Preliminary findings. Fetal Diagn. Ther. 2012, 31, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Lacey, S.R.; Carris, L.A.; Beyer, A.J., 3rd; Azizkhan, R.G. Bladder pressure monitoring significantly enhances care of infants with abdominal wall defects: A prospective clinical study. J. Pediatr. Surg. 1993, 28, 1370–1374; discussion 1374–1375. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.C.; Fonkalsrud, E.W. Improved survival of infants with omphalocele. Am. J. Surg. 1997, 173, 284–287. [Google Scholar] [CrossRef]
- Schuster, S.R. A new method for the staged repair of large omphaloceles. Surg. Gynecol. Obstet. 1967, 125, 837–850. [Google Scholar]
- Kirkpatrick, A.W.; Roberts, D.J.; De Waele, J.; Jaeschke, R.; Malbrain, M.L.; De Keulenaer, B.; Duchesne, J.; Bjorck, M.; Leppaniemi, A.; Ejike, J.C.; et al. Intra-abdominal hypertension and the abdominal compartment syndrome: Updated consensus definitions and clinical practice guidelines from the World Society of the Abdominal Compartment Syndrome. Intensive Care Med. 2013, 39, 1190–1206. [Google Scholar] [CrossRef]
- Bianchi, A.; Dickson, A.P. Elective delayed reduction and no anesthesia: ‘minimal intervention management’ for gastrochisis. J. Pediatr. Surg. 1998, 33, 1338–1340. [Google Scholar] [CrossRef]
- Grabski, D.F.; Hu, Y.; Vavolizza, R.D.; Rasmussen, S.K.; Swanson, J.R.; McGahren, E.D.; Gander, J.W. Sutureless closure: A versatile treatment for the diverse presentations of gastroschisis. J. Perinatol. 2019, 39, 666–672. [Google Scholar] [CrossRef]
- Bruzoni, M.; Jaramillo, J.D.; Dunlap, J.L.; Abrajano, C.; Stack, S.W.; Hintz, S.R.; Hernandez-Boussard, T.; Dutta, S. Sutureless vs Sutured Gastroschisis Closure: A Prospective Randomized Controlled Trial. J. Am. Coll. Surg. 2017, 224, 1091–1096.e1091. [Google Scholar] [CrossRef]
- Sandler, A.; Lawrence, J.; Meehan, J.; Phearman, L.; Soper, R. A “plastic” sutureless abdominal wall closure in gastroschisis. J. Pediatr. Surg. 2004, 39, 738–741. [Google Scholar] [CrossRef]
- Lawrence, L.; Gavens, E.; Reda, B.; Hill, T.; Jester, I.; Lander, A.; Soccorso, G.; Pachl, M.; Gee, O.; Singh, M.; et al. Exomphalos major: Conservative management using Manuka honey dressings and an outreach surgical nursing team. J. Pediatr. Surg. 2021, 56, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, M.; Norris, K.; Uitvlugt, N.; DeCou, J.; Connors, R. Improved outcomes in the treatment of gastroschisis using a preformed silo and delayed repair approach. J. Pediatr. Surg. 2003, 38, 459–464; discussion 459–464. [Google Scholar] [CrossRef] [PubMed]
- Allotey, J.; Davenport, M.; Njere, I.; Charlesworth, P.; Greenough, A.; Ade-Ajayi, N.; Patel, S. Benefit of preformed silos in the management of gastroschisis. Pediatr. Surg. Int. 2007, 23, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.; Abantanga, F.; Amoah, M.; Appeadu-Mensah, W.; Bokhary, Z.; Bvulani, B.; Davies, J.; Miti, S.; Nandi, B.; Nimako, B.; et al. Developing and implementing an interventional bundle to reduce mortality from gastroschisis in low-resource settings. Wellcome Open Res. 2019, 4, 46. [Google Scholar] [CrossRef]
- Philipo, G.S.; Bokhary, Z.M.; Kapapa, M.; Bayyo, N.L.; Nyamuryekung’e, M.K.; Salim, M.; Mboma, L.; Massenga, A.; Michael, L.; Mashara, M.; et al. Improving care and survival of newborns with surgical conditions in Tanzania (TINY Tanzania): A focus on gastroschisis. Pediatr. Surg. Int. 2024, 40, 250. [Google Scholar] [CrossRef]
- Lewis, J.E., Jr.; Kraeger, R.R.; Danis, R.K. Gastroschisis: Ten-year review. Arch. Surg. 1973, 107, 218–222. [Google Scholar] [CrossRef]
- Ein, S.H.; Rubin, S.Z. Gastroschisis: Primary closure or Silon pouch. J. Pediatr. Surg. 1980, 15, 549–552. [Google Scholar] [CrossRef]
- Nakayama, D.K.; Mutich, R.; Motoyama, E.K. Pulmonary dysfunction after primary closure of an abdominal wall defect and its improvement with bronchodilators. Pediatr. Pulmonol. 1992, 12, 174–180. [Google Scholar] [CrossRef]
- Danzer, E.; Hedrick, H.L.; Rintoul, N.E.; Siegle, J.; Adzick, N.S.; Panitch, H.B. Assessment of early pulmonary function abnormalities in giant omphalocele survivors. J. Pediatr. Surg. 2012, 47, 1811–1820. [Google Scholar] [CrossRef]
- van Eijck, F.C.; Hoogeveen, Y.L.; van Weel, C.; Rieu, P.N.; Wijnen, R.M. Minor and giant omphalocele: Long-term outcomes and quality of life. J. Pediatr. Surg. 2009, 44, 1355–1359. [Google Scholar] [CrossRef]
- Zaccara, A.; Iacobelli, B.D.; Calzolari, A.; Turchetta, A.; Orazi, C.; Schingo, P.; Bagolan, P. Cardiopulmonary performances in young children and adolescents born with large abdominal wall defects. J. Pediatr. Surg. 2003, 38, 478–481; discussion 478–481. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberti, P.; Ade-Ajayi, N.; Greenough, A. Respiratory Support Strategies for Surgical Neonates: A Review. Children 2025, 12, 273. https://doi.org/10.3390/children12030273
Alberti P, Ade-Ajayi N, Greenough A. Respiratory Support Strategies for Surgical Neonates: A Review. Children. 2025; 12(3):273. https://doi.org/10.3390/children12030273
Chicago/Turabian StyleAlberti, Piero, Niyi Ade-Ajayi, and Anne Greenough. 2025. "Respiratory Support Strategies for Surgical Neonates: A Review" Children 12, no. 3: 273. https://doi.org/10.3390/children12030273
APA StyleAlberti, P., Ade-Ajayi, N., & Greenough, A. (2025). Respiratory Support Strategies for Surgical Neonates: A Review. Children, 12(3), 273. https://doi.org/10.3390/children12030273