Evolving Cognitive Dysfunction in Children with Neurologically Stable Opsoclonus–Myoclonus Syndrome



Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pang, K.K.; de Sousa, C.; Lang, B.; Pike, M.G. A prospective study of the presentation and management of dancing eye syndrome/opsoclonus–myoclonus syndrome in the United Kingdom. Eur. J. Paediatr. Neurol. 2010, 14, 156–161. [Google Scholar] [CrossRef]
- Gorman, M.P. Update on diagnosis, treatment, and prognosis in opsoclonus–myoclonus–ataxia syndrome. Curr. Opin. Pediatr. 2010, 22, 745–750. [Google Scholar] [CrossRef]
- Berridge, G.; Menassa, D.A.; Moloney, T.; Waters, P.J.; Welding, I.; Thomsen, S.; Zuberi, S.; Fischer, R.; Aricescu, A.R.; Pike, M.; et al. Glutamate receptor δ2 serum antibodies in pediatric opsoclonus myoclonus ataxia syndrome. Neurology 2018, 91, e714–e723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, K.; Jeremy, R.J.; Jenkins, S.; Barkovich, A.J.; Gultekin, S.H.; Kramer, J.; Crittenden, M.; Matthay, K.K. Long-term neurobehavioral outcomes in children with neuroblastoma and opsoclonus-myoclonus-ataxia syndrome: Relationship to MRI findings and anti-neuronal antibodies. J. Pediatr. 2001, 139, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.G.; Davalos-Gonzalez, Y.; Brumm, V.L.; Aller, S.K.; Burger, E.; Turkel, S.B.; Borchert, M.S.; Hollar, S.; Padilla, S. Opsoclonus-ataxia caused by childhood neuroblastoma: Developmental and neurologic sequelae. Pediatrics 2002, 109, 86–98. [Google Scholar] [PubMed]
- Mitchell, W.G.; Brumm, V.L.; Azen, C.G.; Patterson, K.E.; Aller, S.K.; Rodriguez, J. Longitudinal neurodevelopmental evaluation of children with opsoclonus-ataxia. Pediatrics 2005, 116, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Tate, E.D.; Allison, T.J.; Pranzate, M.R.; Verhu, S.J. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J. Pediatr. Oncol. Nurs. 2005, 22, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Pohl, K.; Zuberi, S.M.; de Sousa, C. Outcome and prognostic features in opsoclonus-myoclonus syndrome from infancy to adult life. Pediatrics 2011, 128, e388–e394. [Google Scholar] [CrossRef] [PubMed]
- Anand, G.; Bridge, H.; Rackstraw, P.; Chekroud, A.M.; Yong, J.; Stagg, C.J.; Pike, M. Cerebellar and cortical abnormalities in paediatric opsoclonus-myoclonus syndrome. Dev. Med. Child Neurol. 2015, 57, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranzatelli, M.R.; Travelstead, A.L.; Tate, E.D.; Allison, T.J.; Moticka, E.J.; Franz, D.N.; Nigro, M.A.; Parke, J.T.; Stumpf, D.A.; Verhulst, S.J. B- and T-cell markers in opsoclonus-myoclonus syndrome: Immunophenotyping of CSF lymphocytes. Neurology 2004, 62, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials. Opsoclonus Myoclonus Syndrome/Dancing Eye Syndrome (OMS/DES) in Children with and without Neuroblastoma; Identifier: NCT01868269; National Library of Medicine: Bethesda, MD, USA, 2019.
- Sheridan, A.; Kapur, K.; Pinard, F.; Dietrich Alber, F.; Camposano, S.; Pike, M.G.; Klein, A.; Gorman, M.P. Predictors of Intelligence Quotient (IQ) in a multinational study of 81 patients with pediatric opsoclonus myoclonus ataxia syndrome. Dev. Med. Child Neurol. 2020. In press. [Google Scholar] [CrossRef] [PubMed]
- Ekmekci, O. Pediatric Multiple Sclerosis and Cognition: A Review of Clinical, Neuropsychologic, and Neuroradiologic Features. Behav. Neurol. 2017, 2017, 1463570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, V.; Spencer-Smith, M.; Wood, A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain 2011, 134, 2197–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, Y.; Halperin, J.M.; Newcorn, J.H.; Davey, C.; Fifer, W.P.; Savitz, D.A.; Brooks-Gunn, J. The risk for impaired learning-related abilities in childhood and educational attainment among adults born near-term. J. Pediatr. Psychol. 2009, 34, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Schendel, D.; Bhasin, T.K. Birth weight and gestational age characteristics of children with autism, including a comparison with other developmental disabilities. Pediatrics 2008, 121, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Hillemeier, M.M.; Farkas, G.; Morgan, P.L.; Martin, M.A.; Maczuga, S.A. Disparities in the prevalence of cognitive delay: How early do they appear? Paediatr. Perinat. Epidemiol. 2009, 23, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allotey, J.; Zamora, J.; Cheong-See, F.; Kalidindi, M.; Arroyo-Manzano, D.; Asztalos, E.; van der Post, J.A.M.; Mol, B.W.; Moore, D.; Birtles, D.; et al. Cognitive, motor, behavioural and academic performances of children born preterm: A meta-analysis and systematic review involving 64 061 children. BJOG An Int. J. Obstet. Gynaecol. 2018, 125, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.C.; Reuter-Lorenz, P. The adaptive brain: Aging and neurocognitive scaffolding. Annu. Rev. Psychol. 2009, 60, 173–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A. Slowing down: Age-related neurobiological predictors of processing speed. Front. Neurosci. 2011, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Olsson, I.T.; Perrin, S.; Lundgren, J.; Hjorth, L.; Johanson, A. Long-term cognitive sequelae after pediatric brain tumor related to medical risk factors, age, and sex. Pediatr. Neurol. 2014, 51, 515–521. [Google Scholar] [CrossRef]
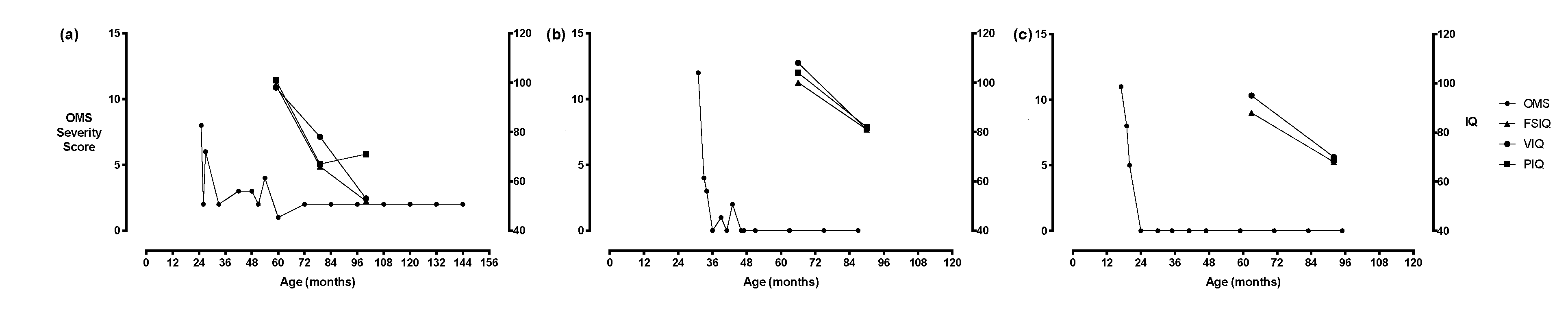

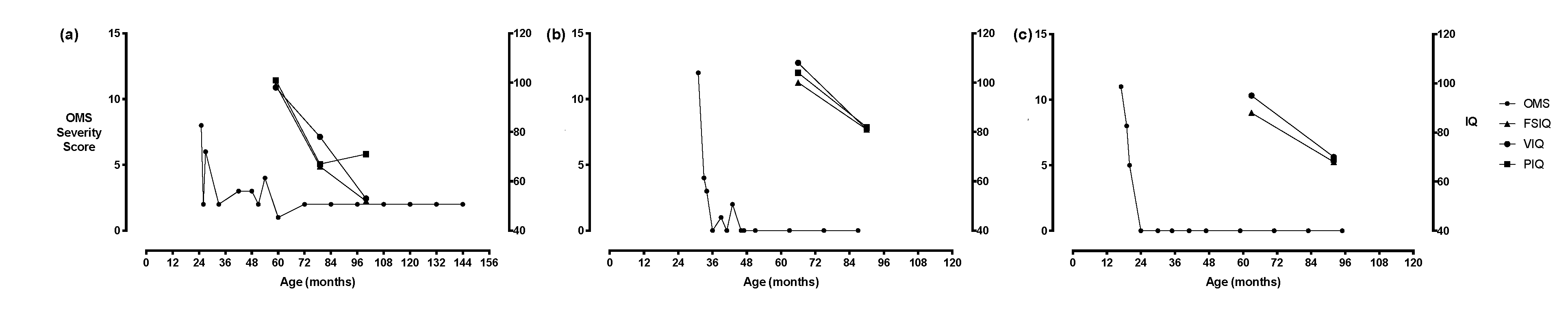{kind=link}
| Case | Gender | Age at Onset | Age at Diagnosis | Clinical Features at Presentation | OMS Score at Diagnosis | Presence of Neuroblastoma | Treatment | Response | Age at Last Follow-Up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 23 months | 25 months | Unsteady gait, abnormal eye movements, opsoclonus, titubation, intention tremor and ataxia | 8/15 | No | Prednisolone IVIG | Good | 13 years |
| 2 | Female | 30 months | 34 months | Unsteady gait, vomiting, opsoclonus and regression in language | 12/15 | No | Dexamethasone | Good | 7 years 6 months |
| 3 | Female | 17 months | 17 months | Limb tremors, ataxia, loss of lower limb and truncal control and loss of speech | 11/15 | Yes | Surgical resection of neuroblastoma Dexamethasone Cyclophosphamide | Good | 7 years 8 months |
| Case | Age | Intellectual Functioning | Other Domains Tested |
|---|---|---|---|
| 1 | 4 years 11 months | FSIQ: 99 VIQ: 98 PIQ: 101 | Attention (selective): – Visuospatial skills: ++ Language (receptive): – Language (expressive): – |
| 6 years 7 months | FSIQ: 66 VIQ: 78 PIQ: 67 | Processing speed: ++ Language (receptive): – Language (expressive): – | |
| 8 years 4 months | FSIQ: 5 VIQ: 53 PIQ: 71 | Attention (working memory): ++ Processing speed: ++ Visuospatial skills: ++ Language (receptive): ++ Language (expressive): ++ Academic (reading): ++ Academic (spelling): ++ | |
| 2 | 5 years 6 months | FSIQ: 100 VIQ: 108 PIQ: 104 | Attention (sustained): – Attention (working memory): – Processing speed: – Visuospatial skills: + Memory (new learning): – Memory (delayed recall): – |
| 7 years 6 months | FSIQ: 81 VIQ: 81 PIQ: 82 | Attention (sustained): ++ Attention (working memory): – Processing speed: – Visuospatial skills: + Language (receptive): – Memory (new learning): – Memory (delayed recall): – Academic (reading): – Academic (spelling): – | |
| 3 | 5 years 3 months | FSIQ: 81 VIQ: 95 | Not tested |
| 7 years 8 months | FSIQ: 68 VIQ: 70 PIQ: 69 | Attention (sustained): ++ Processing speed: ++ Language (receptive): ++ Language (expressive): – Memory (new learning): – Memory (delayed recall): – Academic (reading): – Academic (spelling): – |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goh, E.L.; Scarff, K.; Satariano, S.; Lim, M.; Anand, G. Evolving Cognitive Dysfunction in Children with Neurologically Stable Opsoclonus–Myoclonus Syndrome. Children 2020, 7, 103. https://doi.org/10.3390/children7090103
Goh EL, Scarff K, Satariano S, Lim M, Anand G. Evolving Cognitive Dysfunction in Children with Neurologically Stable Opsoclonus–Myoclonus Syndrome. Children. 2020; 7(9):103. https://doi.org/10.3390/children7090103
Chicago/Turabian StyleGoh, En Lin, Kate Scarff, Stephanie Satariano, Ming Lim, and Geetha Anand. 2020. "Evolving Cognitive Dysfunction in Children with Neurologically Stable Opsoclonus–Myoclonus Syndrome" Children 7, no. 9: 103. https://doi.org/10.3390/children7090103
APA StyleGoh, E. L., Scarff, K., Satariano, S., Lim, M., & Anand, G. (2020). Evolving Cognitive Dysfunction in Children with Neurologically Stable Opsoclonus–Myoclonus Syndrome. Children, 7(9), 103. https://doi.org/10.3390/children7090103