
Characteristics of Clinical Trial Participants with Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet)

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Trial Participation
2.2. Clinical Characteristics
2.3. Sociodemographic Characteristics
2.4. Statistical Analysis
3. Results
4. Discussion
Strengths and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; De Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar] [PubMed]
- Humbertclaude, V.; Hamroun, D.; Bezzou, K.; Bérard, C.; Boespflug-Tanguy, O.; Bommelaer, C.; Campana-Salort, E.; Cances, C.; Chabrol, B.; Commare, M.-C.; et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. Eur. J. Paediatr. Neurol. 2012, 16, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Merino, E.; Bach, J.R. Duchenne Muscular Dystrophy. Am. J. Phys. Med. Rehabil. 2002, 81, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Bach, J.R.; Martínez-González, D. Duchenne Muscular Dystrophy: Continuous Noninvasive Ventilatory Support Prolongs Survival. Respir. Care 2011, 56, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miura, T.; Ishikawa, Y.; Aoyagi, T.; Ogata, H.; Hamada, S.; Minami, R. Duchenne muscular dystrophy: Survival by cardio-respiratory interventions. Neuromuscul. Disord. 2011, 21, 47–51. [Google Scholar] [CrossRef]
- Shieh, P.B. Duchenne muscular dystrophy. Curr. Opin. Neurol. 2015, 28, 542–546. [Google Scholar] [CrossRef]
- Franson, T.; Kinnett, K.; Cripe, T.P. Unique Burdens of Pediatric Clinical Trials in Duchenne Muscular Dystrophy, April 20–21, 2017, Bethesda, Maryland, USA. Ther. Innov. Regul. Sci. 2019, 53, 154–163. [Google Scholar] [CrossRef]
- Hoberman, A.; Shaikh, N.; Bhatnagar, S.; Haralam, M.A.; Kearney, D.H.; Colborn, D.K.; Kienholz, M.L.; Wang, L.; Bunker, C.H.; Keren, R.; et al. Factors That Influence Parental Decisions to Participate in Clinical Research. JAMA Pediatr. 2013, 167, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engster, S.A.; Fascetti, C.; Daw, K.; Reis, E.C. Parent Perceptions of and Preferences for Participation in Child Health Research: Results from a Pediatric Practice-Based Research Network. J. Am. Board Fam. Med. 2019, 32, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peay, H.L.; Biesecker, B.B.; Wilfond, B.S.; Jarecki, J.; Umstead, K.L.; Escolar, D.M.; Tibben, A. Barriers and facilitators to clinical trial participation among parents of children with pediatric neuromuscular disorders. Clin. Trials 2018, 15, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Tanner, A.; Kim, S.-H.; Friedman, D.B.; Foster, C.; Bergeron, C.D. Barriers to Medical Research Participation as Perceived by Clinical Trial Investigators: Communicating with Rural and African American Communities. J. Health Commun. 2014, 20, 88–96. [Google Scholar] [CrossRef]
- Kim, S.-H.; Tanner, A.; Friedman, D.B.; Foster, C.; Bergeron, C.D. Barriers to Clinical Trial Participation: Comparing Perceptions and Knowledge of African American and White South Carolinians. J. Health Commun. 2015, 20, 816–826. [Google Scholar] [CrossRef]
- Kim, S.-H.; Tanner, A.; Friedman, D.B.; Foster, C.; Bergeron, C.D. Barriers to Clinical Trial Participation: A Comparison of Rural and Urban Communities in South Carolina. J. Community Health 2013, 39, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Wendler, D.; Kington, R.; Madans, J.; Van Wye, G.; Christ-Schmidt, H.; Pratt, L.A.; Brawley, O.W.; Gross, C.P.; Emanuel, E. Are Racial and Ethnic Minorities Less Willing to Participate in Health Research? PLoS Med. 2005, 3, e19. [Google Scholar] [CrossRef] [Green Version]
- Vose, J.M. Minority Enrollment to Clinical Trials: Road to Increased Access. Oncology 2021, 35, 107. [Google Scholar] [CrossRef]
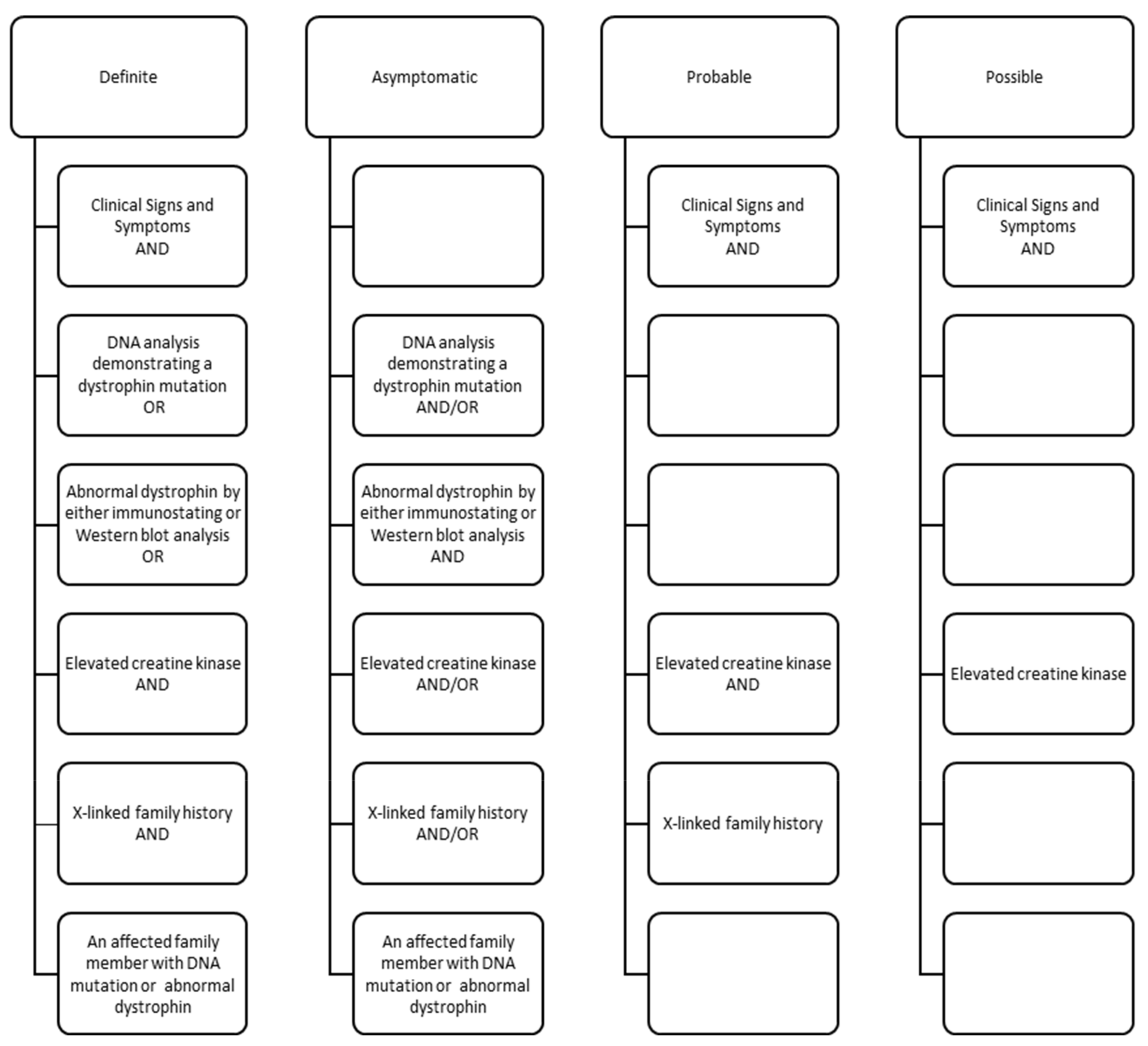
- Mathews, K.D.; Cunniff, C.; Kantamneni, J.R.; Ciafaloni, E.; Miller, T.; Matthews, D.; Cwik, V.; Druschel, C.; Miller, L.; Meaney, F.J.; et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): Case Definition in Surveillance for Childhood-Onset Duchenne/Becker Muscular Dystrophy. J. Child Neurol. 2010, 25, 1098–1102. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.A.; Romitti, P.A.; Cunniff, C.; Druschel, C.; Mathews, K.; Meaney, F.J.; Matthews, D.; Kantamneni, J.; Feng, Z.-F.; Zemblidge, N.; et al. The muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): Surveillance methodology. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Romitti, P.A.; Zhu, Y.; Puzhankara, S.; James, K.A.; Nabukera, S.K.; Zamba, G.K.; Ciafaloni, E.; Cunniff, C.; Druschel, C.M.; Mathews, K.D.; et al. Prevalence of Duchenne and Becker Muscular Dystrophies in the United States. Pediatrics 2015, 135, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, T.N.; Street, N.; Donnelly, J.; Adams, M.M.; Cunniff, C.; Fox, D.J.; Weinert, R.O.; Oleszek, J.; Romitti, P.A.; Westfield, C.P.; et al. Muscular Dystrophy Surveillance, Tracking, and Research Network pilot: Population-based surveillance of major muscular dystrophies at four U.S. sites, 2007–2011. Birth Defects Res. 2018, 110, 1404–1411. [Google Scholar] [CrossRef]
- Wallace, B.; Smith, K.T.; Thomas, S.; Conway, K.M.; Westfield, C.; Andrews, J.G.; Weinert, R.O.; Do, T.Q.N.; Street, N. Characterization of individuals with selected muscular dystrophies from the expanded pilot of the Muscular Dystrophy Surveillance, Tracking and Research Network (MD STARnet) in the United States. Birth Defects Res. 2020, 113, 560–569. [Google Scholar] [CrossRef]
- Zhang, Y.; Mann, J.R.; James, K.A.; McDermott, S.; Conway, K.M.; Paramsothy, P.; Smith, T.; Cai, B.; Starnet, T.M. Duchenne and Becker Muscular Dystrophies’ Prevalence in MD STARnet Surveillance Sites: An Examination of Racial and Ethnic Differences. Neuroepidemiology 2021, 55, 47–55. [Google Scholar] [CrossRef]
- Muscular Dystrophy Association. Highlights of the MDA U.S. Neuromuscular Disease Registry (2013–2016). Available online: https://www.mda.org/sites/default/files/MDA%20Registry%20Report%20Highlights_Digital_final.pdf (accessed on 18 June 2021).
- Heller, C.; Balls-Berry, J.E.; Nery, J.D.; Erwin, P.J.; Littleton, D.; Kim, M.; Kuo, W.P. Strategies addressing barriers to clinical trial enrollment of underrepresented populations: A systematic review. Contemp. Clin. Trials 2014, 39, 169–182. [Google Scholar] [CrossRef]
- Bendixen, R.M.; Morgenroth, L.P.; Clinard, K.L. Engaging Participants in Rare Disease Research: A Qualitative Study of Duchenne Muscular Dystrophy. Clin. Ther. 2016, 38, 1474–1484.e2. [Google Scholar] [CrossRef]
- Cunningham-Erves, J.; Villalta-Gil, V.; Wallston, K.A.; Boyer, A.P.; Wilkins, C.H. Racial differences in two measures of trust in biomedical research. J. Clin. Transl. Sci. 2019, 3, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evelyn, B.; Toigo, T.; Banks, D.; Pohl, D.; Gray, K.; Robins, B.; Ernat, J. Participation of racial/ethnic groups in clinical trials and race-related labeling: A review of new molecular entities approved 1995–1999. J. Natl. Med Assoc. 2001, 93, 18S–24S. [Google Scholar] [PubMed]
- Enhancing the Diversity of Clinical Trial Populations: Eligibility Criteria, Enrollment Practices, and Trial Designs Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/enhancing-diversity-clinical-trial-populations-eligibility-criteria-enrollment-practices-and-trial (accessed on 18 June 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | n (%) |
|---|---|
| Total Sample | 358 |
| Clinical Trial Eligible Mutations, All Individuals | |
| Exon-Skippable (exon 51) Deletions, n (% total) | 34 (9.5) |
| Nonsense (premature stop codon) Mutations, n (% total) | 37 (10.3) |
| Clinical Trial Participation, n (% total) 1 | 64 (17.9) |
| Clinical Trial Checkbox | 59 (16.5) |
| Corticosteroid Clinical Trial | 15 (4.2) |
| Clinical Trial Medication (non-corticosteroid) | 33 (9.2) |
| Clinical Trial Medication, n (% total medications) 2 | 33 |
| Tadalafil | 9 (27.3) |
| Ataluren (PTC124) | 9 (27.3) |
| Idebenone | 4 (12.1) |
| Drisapersen (GSK2402968) | 3 (9.1) |
| CAT-1004 | 2 (6.1) |
| IGF-1 | 2 (6.1) |
| Domegrozumab (PF-06252616) | 1 (3.0) |
| Eplerenone | 1 (3.0) |
| Eteplirsen | 1 (3.0) |
| Unspecified | 1 (3.0) |
| Characteristics | In Clinical Trial | Not in Clinical Trial | p-Value |
|---|---|---|---|
| Total | 64 | 294 | |
| MD STARnet Site, n (row %) | 0.0118 1 | ||
| Site A | 15 (17.0) | 73 (83.0) | |
| Site B | 10 (17.9) | 46 (82.1) | |
| Site C | 10 (17.0) | 49 (83.0) | |
| Site D | 12 (26.0) | 34 (74.0) | |
| Site E | 2 (3.7) | 52 (96.3) | |
| Site F | 15 (27.3) | 40 (72.7) | |
| Child characteristics | |||
| Child Race/Ethnicity, n (row %) | 0.0486 1 | ||
| non-Hispanic white | 52 (21.4) | 191 (78.6) | |
| non-Hispanic black | 1 (4.0) | 24 (96.0) | |
| non-Hispanic other | 2 (15.4) | 11 (84.6) | |
| Hispanic or Latino/Latina | 7 (10.8) | 58 (89.2) | |
| Unknown | 2 | 10 | |
| County-level sociodemographics | |||
| Race/ethnicity (2015), Mean (95% CL) 3 | |||
| non-Hispanic white alone | 65.5 (61.1, 70.0) | 62.4 (60.6, 64.3) | 0.1019 2 |
| non-Hispanic black alone | 8.1 (5.5, 10.7) | 11.6 (10.1, 13.1) | 0.0115 2 |
| non-Hispanic other or combined | 7.4 (6.6, 8.1) | 7.4 (7.1, 7.7) | 0.4596 2 |
| Hispanic | 19.0 (16.4, 21.7) | 18.6 (17.2, 20.0) | 0.1383 2 |
| Education (2014–2018), Mean (95% CL) 4 | |||
| Less than HS | 9.6 (8.9, 10.2) | 10.2 (9.8, 10.7) | 0.3228 2 |
| HS | 26.1 (24.4, 27.9) | 26.3 (25.5, 27.0) | 0.4385 2 |
| Some college | 31.7 (30.7, 32.7) | 31.5 (31.0, 31.9) | 0.3416 2 |
| Bachelor’s degree | 32.6 (30.1, 35.2) | 32.0 (30.8, 33.2) | 0.4536 2 |
| Economic indicators 5 | |||
| Household income (2015), median (95% CL) | $58,367 ($56,067, $60,667) | $57,148 ($55,920, $58,376) | 0.1631 2 |
| Poverty (2015), median percent of population (95% CL) | 17.0 (15.6, 18.4) | 18.0 (17.3, 18.6) | 0.1115 2 |
| Rural-Urban Continuum Codes, n (row %) 6 | 0.4656 1 | ||
| Metropolitan area—1 million population or more | 29 (22.3) | 101 (77.7) | |
| Metropolitan area—250,000 to 1 million population | 20 (14.3) | 120 (85.7) | |
| Metropolitan area—fewer than 250,000 population | 3 (12.5) | 21 (87.5) | |
| Nonmetropolitan urban area or rural area adjacent to metropolitan area | 8 (20.5) | 31 (79.5) | |
| Nonmetropolitan urban area or rural area not adjacent to metropolitan area | 3 (15.0) | 17 (85.0) |
| Characteristics | In Clinical Trial | Not in Clinical Trial | p-Value |
|---|---|---|---|
| Total | 64 | 294 | |
| Ages (years), Mean (95% confidence limits) | |||
| First visit 1 | 5.1 (4.4, 5.7) | 4.9 (4.5, 5.2) | 0.2586 2 |
| Last visit | 9.6 (8.9, 10.3) | 9.3 (8.9, 9.7) | 0.4015 2 |
| Ambulation status, n (row %) | 0.0225 3 | ||
| Not walking | 11 (10.6) | 93 (89.4) | |
| Walking | 53 (20.9) | 201 (79.1) | |
| Non-trial corticosteroid use, n (row %) | <0.0001 3 | ||
| No | 5 (4.8) | 100 (95.2) | |
| Yes | 59 (23.3) | 194 (76.7) | |
| Family history, n (row %) | 0. 3832 3 | ||
| Definite | 18 (14.9) | 103 (85.1) | |
| Suspected | 0 (0.0) | 8 (100.0) | |
| No known | 8 (22.2) | 28 (77.8) | |
| Unknown | 38 (19.7) | 155 (80.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathews, K.D.; Conway, K.M.; Gedlinske, A.M.; Johnson, N.; Street, N.; Butterfield, R.J.; Hung, M.; Ciafaloni, E.; Romitti, P.A. Characteristics of Clinical Trial Participants with Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). Children 2021, 8, 835. https://doi.org/10.3390/children8100835
Mathews KD, Conway KM, Gedlinske AM, Johnson N, Street N, Butterfield RJ, Hung M, Ciafaloni E, Romitti PA. Characteristics of Clinical Trial Participants with Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). Children. 2021; 8(10):835. https://doi.org/10.3390/children8100835
Chicago/Turabian StyleMathews, Katherine D., Kristin M. Conway, Amber M. Gedlinske, Nicholas Johnson, Natalie Street, Russell J. Butterfield, Man Hung, Emma Ciafaloni, and Paul A. Romitti. 2021. "Characteristics of Clinical Trial Participants with Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet)" Children 8, no. 10: 835. https://doi.org/10.3390/children8100835
APA StyleMathews, K. D., Conway, K. M., Gedlinske, A. M., Johnson, N., Street, N., Butterfield, R. J., Hung, M., Ciafaloni, E., & Romitti, P. A. (2021). Characteristics of Clinical Trial Participants with Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). Children, 8(10), 835. https://doi.org/10.3390/children8100835