Primary Immunodeficiency Disease Mimicking Pediatric Bechet’s Disease


Abstract
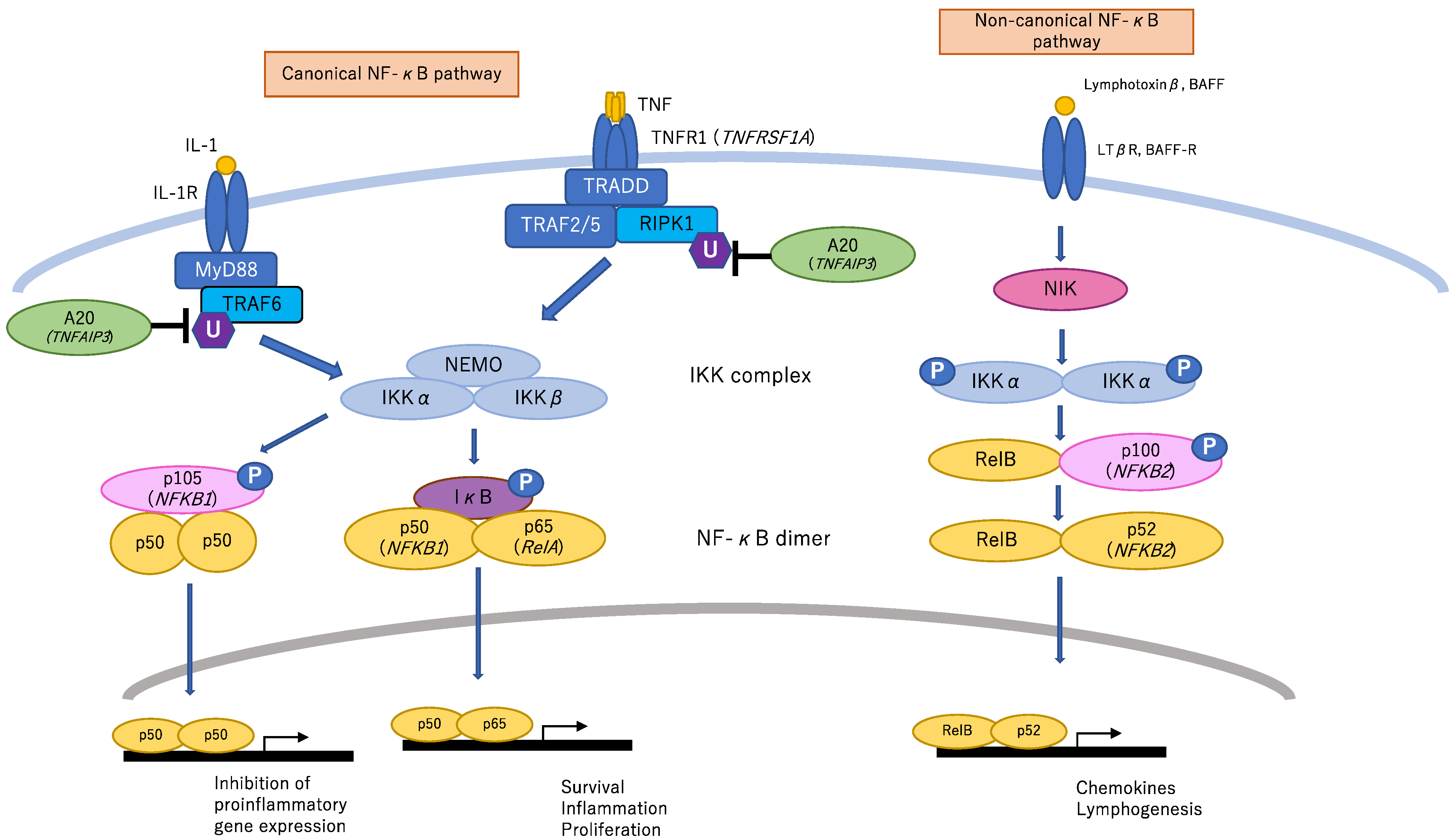
:1. Introduction
2. NF-κB-Related Genes
2.1. TNFAIP3
2.2. NF-κB Essential Modulator (NEMO, also Called IKBKG)
2.3. RELA
2.4. NFKB1
2.5. TNF Receptor Superfamily Member 1A (TNFRSF1A)
3. Other Genes
3.1. DNA Ligase 4 (LIG4)
3.2. WDR1
3.3. Neutrophil Cytosolic Factor 1 (NCF1)
3.4. Signal Transducer and Activator of Transcription (STAT) 1
3.5. Adenosine Deaminase (ADA) 2 (CECR1)
3.6. Mediterranean Fever (MEFV)
3.7. AP1S3
3.8. LYN
3.9. Trisomy 8
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Behcet, H. Uber rezidivierende, aphthose, durch ein Virus verursachte Geschwure am Mund, am Auge und an den Genitalian. Derm. Wochensch 1937, 105, 1152–1157. [Google Scholar]
- Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet 1990, 335, 1078–1080.
- Koné-Paut, I.; Shahram, F.; Darce-Bello, M.; Cantarini, L.; Cimaz, R.; Gattorno, M.; Anton, J.; Hofer, M.; Chkirate, B.; Bouayed, K.; et al. Consensus classification criteria for paediatric Behçet’s disease from a prospective observational cohort: PEDBD. Ann Rheum. Dis. 2016, 75, 958–964. [Google Scholar]
- Ohno, S.; Asanuma, T.; Sugiura, S.; Wakisaka, A.; Aizawa, M.; Itakura, K. HLA-Bw51 and Behçet’s disease. JAMA 1978, 240, 529. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, N.; Meguro, A.; Ota, M.; Ohno, S.; Shiota, T.; Kawagoe, T.; Ito, N.; Kera, J.; Okada, E.; Yatsu, K.; et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet’s disease susceptibility loci. Nat. Genet. 2010, 42, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Remmers, E.F.; Cosan, F.; Kirino, Y.; Ombrello, M.J.; Abaci, N.; Satorius, C.; Le, J.M.; Yang, B.; Korman, B.D.; Cakiris, A.; et al. Genome-wide association study identifies variants in the MHC class, I.; IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat. Genet. 2010, 42, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirino, Y.; Bertsias, G.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Sacli, F.S.; Erer, B.; Inoko, H.; et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat. Genet. 2013, 45, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirino, Y.; Zhou, Q.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Ugurlu, S.; Erer, B.; Abaci, N.; et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behcet disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8134–8139. [Google Scholar] [CrossRef] [Green Version]
- Kappen, J.H.; Medina-Gomez, C.; van Hagen, P.M.; Stolk, L.; Estrada, K.; Rivadeneira, F.; Uitterlinden, A.G.; Stanford, M.R.; Ben-Chetrit, E.; Wallace, G.R.; et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behçet disease. PLoS ONE 2015, 10, e0119085. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Mizuki, N.; Meguro, A.; Ombrello, M.J.; Kirino, Y.; Satorius, C.; Le, J.; Blake, M.; Erer, B.; Kawagoe, T.; et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet’s disease susceptibility. Nat. Genet. 2017, 49, 438–443. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Omoyinmi, E.; Standing, A.; Pain, C.E.; Booth, C.; D’Arco, F.; Gilmour, K.; Buckland, M.; Eleftheriou, D.; Brogan, P.A. Monogenic mimics of Behcet’s disease in the young. Rheumatology (Oxford) 2019, 58, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Pain, C.E. Juvenile-onset Behcet’s syndrome and mimics. Clin. Immunol. 2020, 214, 108381. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perazzio, S.F.; Allenspach, E.J.; Eklund, K.K.; Varjosalo, M.; Shinohara, M.M.; Torgerson, T.R.; Seppänen, M.R.J. Behçet disease (BD) and BD-like clinical phenotypes: NF-κB pathway in mucosal ulcerating diseases. Scand. J. Immunol. 2020, 92, e12973. [Google Scholar] [CrossRef]
- Taskiran, E.Z.; Sonmez, H.E.; Kosukcu, C.; Tavukcuoglu, E.; Yazici, G.; Esendagli, G.; Batu, E.D.; Kiper, P.O.S.; Bilginer, Y.; Alikasifoglu, M.; et al. A Novel Missense LIG4 Mutation in a Patient with a Phenotype Mimicking Behçet’s Disease. J. Clin. Immunol. 2019, 39, 99–105. [Google Scholar] [CrossRef]
- Badran, Y.R.; Dedeoglu, F.; Leyva Castillo, J.M.; Bainter, W.; Ohsumi, T.K.; Bousvaros, A.; Goldsmith, J.D.; Geha, R.S.; Chou, J. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J. Exp. Med. 2017, 214, 1937–1947. [Google Scholar] [CrossRef]
- Adeeb, F.; Dorris, E.R.; Morgan, N.E.; Lawless, D.; Maqsood, A.; Ng, W.L.; Killeen, O.; Cummins, E.P.; Taylor, C.T.; Savic, S.; et al. A novel RELA truncating mutation in familial Behcet’s Disease-like mucocutaneous ulcerative condition. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Takada, H.; Nomura, A.; Ishimura, M.; Ichiyama, M.; Ohga, S.; Hara, T. NEMO mutation as a cause of familial occurrence of Behçet’s disease in female patients. Clin. Genet. 2010, 78, 575–579. [Google Scholar] [CrossRef]
- Klemann, C.; Pannicke, U.; Morris-Rosendahl, D.J.; Vlantis, K.; Rizzi, M.; Uhlig, H.; Vraetz, T.; Speckmann, C.; Strahm, B.; Pasparakis, M.; et al. Transplantation from a symptomatic carrier sister restores host defenses but does not prevent colitis in NEMO deficiency. Clin. Immunol. 2016, 164, 52–56. [Google Scholar] [CrossRef]
- Baldini, L.; Di Sabatino, F.; Bodrero, E.; Dellepiane, M.; Covizzi, C.; La Selva, R.; Montin, D.; Licciardi, F. NeMO mutations: A rare cause of monogenic Behcet-like disease. Rheumatology (Oxford) 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaustio, M.; Haapaniemi, E.; Goos, H.; Hautala, T.; Park, G.; Syrjanen, J.; Einarsdottir, E.; Sahu, B.; Kilpinen, S.; Rounioja, S.; et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J. Allergy Clin. Immunol. 2017, 140, 782–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, I.; Dulek, D.E.; Creech, C.B.; Graham, T.B.; Williams, J.V. Chronic granulomatous disease masquerading as Behçet disease: A case report and review of the literature. Pediatric Infect. Dis. J. 2012, 31, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, D.; Bloomfield, F.J.; Lenehan, T.; Griffin, M.; Feighery, C.; McCann, S.R. Chronic granulomatous disease presenting as an oculomucocutaneous syndrome mimicking Behçet’s syndrome. Postgrad. Med. J. 1986, 62, 489–491. [Google Scholar] [CrossRef] [Green Version]
- van Well, G.T.J.; Kant, B.; van Nistelrooij, A.; Sirma Ekmekci, S.; Henriet, S.V.; Hoppenreijs, E.; van Deuren, M.; van Montfrans, J.; Nierkens, S.; Gül, A.; et al. Phenotypic variability including Behçet’s disease-like manifestations in DADA2 patients due to a homozygous c.973-2A>G splice site mutation. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S121), 142–146. [Google Scholar]
- Kadowaki, T.; Ohnishi, H.; Kawamoto, N.; Hori, T.; Nishimura, K.; Kobayashi, C.; Shigemura, T.; Ogata, S.; Inoue, Y.; Kawai, T.; et al. Haploinsufficiency of A20 causes autoinflammatory and autoimmune disorders. J. Allergy Clin. Immunol. 2018, 141, 1485–1488.e11. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.P.; Xu, X.S.; Zhou, Q.; Deuitch, N.; Lu, M.P. Haploinsufficiency of A20 (HA20): Updates on the genetics, phenotype, pathogenesis and treatment. World J. Pediatrics 2020, 16, 575–584. [Google Scholar] [CrossRef]
- Tsuchida, N.; Kirino, Y.; Soejima, Y.; Onodera, M.; Arai, K.; Tamura, E.; Ishikawa, T.; Kawai, T.; Uchiyama, T.; Nomura, S.; et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease. Arthritis Res. Ther. 2019, 21, 137. [Google Scholar] [CrossRef] [Green Version]
- Koguchi-Yoshioka, H.; Inokuma, D.; Kanda, M.; Kondo, M.; Kikuchi, K.; Shimizu, S. Behcet’s disease-like symptoms associated with myelodysplastic syndrome with trisomy 8: A case report and review of the literature. Acta Derm.-Venereol. 2014, 94, 355–356. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Ma, H.F.; Luo, D.; Cai, J.F.; Zou, J.; Guan, J.L. High Incidence of Gastrointestinal Ulceration and Cytogenetic Aberration of Trisomy 8 as Typical Features of Behcet’s Disease Associated with Myelodysplastic Syndrome: A Series of 16 Consecutive Chinese Patients from the Shanghai Behcet’s Disease Database and Comparison with the Literature. Biomed. Res. Int. 2018, 2018, 8535091. [Google Scholar]
- Zonana, J.; Elder, M.E.; Schneider, L.C.; Orlow, S.J.; Moss, C.; Golabi, M.; Shapira, S.K.; Farndon, P.A.; Wara, D.W.; Emmal, S.A.; et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am. J. Hum. Genet. 2000, 67, 1555–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrod, A.E. Peculiar pigmentation of the skin in an infant. Trans. Clin. Soc. Lond. 1906, 39, 216. [Google Scholar]
- de Jesus, A.A.; Hou, Y.; Brooks, S.; Malle, L.; Biancotto, A.; Huang, Y.; Calvo, K.R.; Marrero, B.; Moir, S.; Oler, A.J.; et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J. Clin. Investig. 2020, 130, 1669–1682. [Google Scholar] [CrossRef] [Green Version]
- Nijenhuis, T.; Klasen, I.; Weemaes, C.M.; Preijers, F.; de Vries, E.; van der Meer, J.W. Common variable immunodeficiency (CVID) in a family: An autosomal dominant mode of inheritance. Neth. J. Med. 2001, 59, 134–139. [Google Scholar] [CrossRef]
- Yalcin, B.; Atakan, N.; Alli, N. The functional role of nuclear factor kappa-kappaB1 -94 ins/del ATTG promotor gene polymorphism in Behcet’s disease: An exploratory study. Clin. Exp. Dermatol. 2008, 33, 629–633. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Plowman, P.N.; Bridges, B.A.; Arlett, C.F.; Hinney, A.; Kingston, J.E. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br. J. Radiol. 1990, 63, 624–628. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Plantinga, T.S.; Hoischen, A.; Smeekens, S.P.; Joosten, L.A.; Gilissen, C.; Arts, P.; Rosentul, D.C.; Carmichael, A.J.; Smits-van der Graaf, C.A.; et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 2011, 365, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Navon Elkan, P.; Pierce, S.B.; Segel, R.; Walsh, T.; Barash, J.; Padeh, S.; Zlotogorski, A.; Berkun, Y.; Press, J.J.; Mukamel, M.; et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N. Engl. J. Med. 2014, 370, 921–931. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [Green Version]
- International FMF Consortium: Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 1997, 90, 797–807. [CrossRef]
- French, F.M.F. Consortium: A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar]
- Ishikawa, H.; Shindo, A.; Ii, Y.; Sakano, S.; Asahi, M.; Matsuura, K.; Kishida, D.; Umino, M.; Maeda, M.; Tomimoto, H. Vertebral artery dissection associated with familial Mediterranean fever and Behcet’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Mei, D.; Zhang, L.; Wei, W. Src Family Protein Kinase Controls the Fate of B Cells in Autoimmune Diseases. Inflammation 2020. [Google Scholar] [CrossRef]
- Ban, T.; Sato, G.R.; Nishiyama, A.; Akiyama, A.; Takasuna, M.; Umehara, M.; Suzuki, S.; Ichino, M.; Matsunaga, S.; Kimura, A.; et al. Lyn Kinase Suppresses the Transcriptional Activity of IRF5 in the TLR-MyD88 Pathway to Restrain the Development of Autoimmunity. Immunity 2016, 45, 319–332. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, A.; Montealegre, G.; Liu, Y.; Marrero, B.; Kuehn, H.; Calvo, K.; Rosenzweig, S.; Fleisher, T.; Lee, R.; Brundidge, A.; et al. A de novo nonsense mutation in the tyrosine kinase lyn in a patient with an early onset autoinflammatory phenotype. Pediatric Rheumatol. 2014, 12 (Suppl. S1), O25. [Google Scholar] [CrossRef] [Green Version]
- Kalpana Manthiram, A.D.-F.; Deborah, B.; Amanda, O.; Karyl, B.; Tina, R.; Anne, J. Trisomy 8-associated autoinflammatory disease (TRIAD) is characterized by increased monocyte activation. Pediatric Rheumatol. 2019, 17, 13. [Google Scholar]

{kind=link}
| Classification | Gene/Chromosome | Cytogenetic Location | Disease | Inheritance | Common Features | Reference Number * | |
|---|---|---|---|---|---|---|---|
| Classification of Inborn Errors of Immunity from the IUIS | IDs affecting cellular and humoral immunity | LIG4 | 13q33.3 | LIG4 deficiency | AR | Combined immunodeficiency, microcephaly, abnormal facial features, sensitivity to ionizing radiation, growth failure, susceptibility to malignancy | [16] |
| RELA | 11q13.1 | RelA haploinsufficiency | AD | Chronic mucocutaneous ulceration | [17,18] | ||
| Combined ID with associated or syndromic features | NEMO (IKBKG) | Xq28 | EDA-ID | XLR | Recurrent severe infections caused by immunodeficiency, ectodermal dysplasia, lymphedema, osteopetrosis | [19,20,21] | |
| IP | XLD | Variable abnormalities of skin, hair, nails, teeth, eyes, and central nervous system | |||||
| NEMO- NDAS | XLR/ XLD | Panniculitis, chorioretinitis, progressive B cell lymphopenia, hypogammaglobulinemia | |||||
| Predominantly antibody deficiencies | NFKB1 | 4q24 | NFKB1 deficiency | AD | Recurrent sinopulmonary infections, COPD, autoimmune features | [22] | |
| Congenital defects of phagocyte number or function | WDR1 | 4q16.1 | PFIT | AR | Recurrent respiratory infections, stomatitis, cutaneous infections | [12] | |
| NCF1 | 7q11.23 | CGD | AR | Recurrent infections, abscesses, lymphadenopathy, granulomatous formation, enteritis | [12,23,24] | ||
| Defects in intrinsic and innate immunity | STAT1 | 2q32.2 | ID 31A | AD LOF | Mycobacterial infections | [12] | |
| ID 31B | AR LOF | Severe mycobacterial and viral infections | |||||
| ID 31C | AD GOF | Chronic mucocutaneous candidiasis, recurrent severe infections, enteropathy, autoimmune disorders | |||||
| Autoinflammatory disorders | ADA2 | 22q11.1 | DADA2 | AR LOF | Skin ulceration, recurrent strokes affecting the small vessels of the brain, fever, polyarteritis nodosa | [25] | |
| MEFV | 16p13.3 | FMF | AR LOF/AD | Recurrent attacks of fever, inflammation in the peritoneum, synovium, or pleura, accompanied by pain | [12] | ||
| TNFRSF1A | 12p13.31 | TRAPS | AD | Recurrent fever, localized myalgia, painful erythema, arthralgia, conjunctivitis, serositis | [12] | ||
| TNFAIP3 | 6q23.3 | HA20 | AD LOF | Periodic fever, recurrent aphthous stomatitis, genital ulceration, intestinal symptoms, skin rash, polyarthritis, autoimmune disorders | [12,13,26,27,28] | ||
| AP1S3 | 2q36.1 | Pustular psoriasis | AR | High-grade fever, generalized rash, disseminated pustules | [12] | ||
| Others | LYN | 8q12.1 | LYN-associated AID | AD | Hepatosplenomegaly, purpuric skin rash, periorbital erythema, testicular swelling, positive autoantibodies | [12] | |
| chromosome 8 | Trisomy 8 | Mental retardation, joint contractures, deep palmar and plantar creases, corpus callosum agenesis, skeletal and renal anomalies | [29,30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiraki, M.; Kadowaki, S.; Kadowaki, T.; Kawamoto, N.; Ohnishi, H. Primary Immunodeficiency Disease Mimicking Pediatric Bechet’s Disease. Children 2021, 8, 75. https://doi.org/10.3390/children8020075
Shiraki M, Kadowaki S, Kadowaki T, Kawamoto N, Ohnishi H. Primary Immunodeficiency Disease Mimicking Pediatric Bechet’s Disease. Children. 2021; 8(2):75. https://doi.org/10.3390/children8020075
Chicago/Turabian StyleShiraki, Mayuka, Saori Kadowaki, Tomonori Kadowaki, Norio Kawamoto, and Hidenori Ohnishi. 2021. "Primary Immunodeficiency Disease Mimicking Pediatric Bechet’s Disease" Children 8, no. 2: 75. https://doi.org/10.3390/children8020075