1. Introduction
Wolf–Hirschhorn syndrome (WHS) is a rare contiguous gene deletion syndrome (prevalence of 1:20,000–50,000 births, with a female to male ratio of 2:1 induced by the absence of the distal portion of the short arm of chromosome 4 [
1,
2]. WHS presents a characteristic phenotype that includes intrauterine growth retardation and later on short stature, low weight, hypotonia, intellectual disability, epilepsy, and specific craniofacial dysmorphism (“Greek warrior helmet”). Other associated clinical manifestations include skeletal anomalies, congenital heart defects (septal defects and pulmonary stenosis), eye abnormalities, hearing loss, genitourinary tract defects, and immunological disorders [
3]. The suggestive craniofacial features include wide nasal bridge continuing to the forehead, high anterior hairline with prominent glabella, highly arched eyebrows, widely spaced eyes, epicanthus, short philtrum, downturned corners of the month and micrognathia. Most of the patients have microcephaly and poorly formed ears (lobeless pinnae, underdeveloped or absent cartilage) as well as low-set or posteriorly angulated with pits/tags [
3]. The genetic defect is usually a partial deletion of the distal short arm of chromosome 4, but WHS phenotype could be generated also by complex chromosomal rearrangements, like translocations or ring chromosomes. The unbalanced translocations can be
de novo or inherited from a parent with a balanced rearrangement. The most frequently observed translocations are: (1) involving a rearrangement t(4p;8p), but t(4p;7p), t(4p;11p), t(4p;20q), t(4p;21q) and t(4p;12p, (2) inverted duplications associated with terminal deletions on the same 4p arm or 3, unbalanced pericentric inversions are cited [
3,
4]. There is a great variability in the extent of the deletion, ranging from less than 2 Megabases, (Mb) up to 30 Mb. There are three different categories of WHS phenotypes based on the 4p deletion size: a small deletion (not exceeding 3.5 Mb) which is usually associated with a mild phenotype (clinical manifestations are limited to the typical facial dysmorphism, growth delay, mild intellectual disability, and seizures). The large deletion (5 to 18 Mb) is the most frequent and it is associated with more severe manifestations including hypotonia, severe growth delay, significant neurodevelopmental impairment, and major malformations on top of the ones from the mild type, whereas the very large deletion (exceeding 22–25 Mb) is characterized by the most severe phenotype and is frequently hard to recognize as WHS [
4]. The genomic defect and the pattern of seizures are the most important prognostic factors for the neurodevelopmental issues [
4]. The WHS minimal critical region (MCR) is located approximately 2 Mb from the telomere 4p, within band 4p16.3 [
5] (a region of 200–750 kilobases (kb) rich in genes—
WHSC1, LETM1, NSD2, CPLX1, PIGG being usually deleted) [
6,
7]. The diagnosis requires clinical examination and genetic analysis; deletions greater than 5 MB can be identified by karyotype, but for smaller deletions (microdeletions) molecular diagnostic—CMA, MLPA, fluorescent in situ hybridization (FISH)— is necessary. Standard chromosomal analysis (routine and high resolution) is able to detect about 50–60% of the deletions, whereas FISH, using a
WHSCR probe, detects more than 95%. Chromosomal microarray (CMA) detects all currently known deletions of the
WHSCR and can determine if the deletion is pure or part of a more complex rearrangement. Standard cytogenetics may accompany the CMA to characterize any complex aberration if present. MLPA is a multiplex PCR assay that uses up to 40 probes, each specific for a different DNA sequence [
8]. In recent years, MLPA has become a widely used technique in laboratories performing genetics testing for the molecular diagnosis of several diseases. MLPA is a high throughput analysis and allows a comprehensive identification of gene dosages with relatively low cost equipment and reagents [
9]. A combined MLPA(combination of specific kits for microdeletion screening, followed up with specific kits for microdeletion confirmation) testing can be used to detect microdeletions that may not be identified by FISH. As there are no specific treatments available, supportive management can be useful considering WHS has a high mortality rate (30% within the first two years of life). The most common complications leading to death are respiratory tract infections and congenital heart diseases [
1].
The aim of our study was to identify rare specific characteristics of WHS, to determine if there are major differences based on the size of the deletions and whether there is a correlation between deletion size and phenotypic severity, as well as trying to establish MLPA as a reliable tool for the diagnosis oh WHS.
3. Results
3.1. Small Deletions (<3 Mb)
CASE 1
Case 1 is a 13-year-old boy, first child of a young, unrelated, apparently healthy couple, with no similar cases in the family. The pregnancy was uneventful, fetal ultrasound scan (36 weeks of amenorrhea (WA) revealed a corpus callosum agenesis. The birth was at full term, by cesarean section, (weight (Wt) = 3600 g, height (Ht) = 52 cm, occipital-frontal circumference (OFC) = 36.5 cm, Apgar score 8). Postnatal development was severely delayed (raised head at 18months (Mo), sat without support at 2 years (Y), no speech at age 13Y); the patient has recurrent respiratory infections. Physical evaluation at 13 years and 7 months (Wt: −0.89 standard deviation (SD), Ht: −2.39 SD, OFC: −1.23 SD) revealed: dysmorphic face, defect of lacrimal system, a small palate defect, pectus carinatum, diastasis recti, hypospadias, undescended testes, sacral sinus, delayed tooth eruption, spastic quadriplegia/tetraparesis and severe intellectual disability. MRI showed agenesis of corpus callosum(ACC) and mild dilatation of ventricular system. Other malformations found in this case include: cardiac defects (atrial septal defect (ASD)) and bilateral optic atrophy. Karyotype was 46,XY and MLPA confirmed a ~2 Mb deletion in WHS region.
CASE 2
The second case, a 4-year-old girl, is the only child of non-related parents. The pregnancy evolved with a threatening miscarriage and polyhydramnios. The child was born at term, by cesarean section (Apgar score 9, Wt = 2500 g, Ht = 48 cm, OFC = 31.5 cm). Postnatal development was delayed. Jacksonian seizures have been noted in the right half of the body. Physical examination revealed small size (Wt: −3.2 SD, Ht: −4.15 SD), microcephaly (OFC: −4.66 SD), dysmorphic face, congenital cataract, dental anomalies, sacral sinus, two tuberous hemangiomas, hypotonia. The patient also has ASD, ventricular septal defect (VSD) and patent ductus arteriosus (PDA). Karyotype was 46,XX and MLPA P096 Probemix confirmed a ~2 Mb deletion in WHS region.
CASE 3
Case 3 is a 2-year-old boy, second child of an unrelated, apparently healthy couple, with no similar cases in the family. Pregnancy evolved with intrauterine growth retardation. The child was born at 36 weeks, by cesarean section (Wt = 1400 g, Ht = 30 cm, OFC = 28 cm). Postnatal development is delayed. The last evaluation (1 Y 6 Mo) revealed small size (Wt: −7.09 SD, Ht: −5.7SD), microcephaly (OFC: −4.81 SD), characteristic dysmorphic face (
Figure 1).
He also presents associated brachydactyly, bilateral transverse palmar crease, hypospadias, hypotonia and severe developmental delay. Imagistic investigations identified: ASD, pulmonary artery stenosis, hypoplastic corpus callosum. Karyotype was 46,XY,r(4)(p15.1q35)/46,XY,−4,+mar/47,XY,r(4)(p15.1q35),+mar/46,XY,der(4),+mar (
Figure 2) and MLPA P358 Probemix confirmed a ~2.85 Mb deletion in WHS region (
Figure 3 and P264 Probemix identified a deletion on 4q.
CASE 4
Case 4 is a 3-year-old male with severe developmental delay and multiple congenital anomalies. He is the first child of a young, unrelated couple. During the pregnancy (28 WA) an 8 mm cyst was identified in cavum vergae. At birth he presented respiratory distress (Apgar score 8, Wt = 2600 g, Ht = 49 cm, OFC = 33 cm). Postnatal development is delayed (cannot raise head, sit without help, or speak) and was characterized by seizures and recurrent respiratory infections. Last evaluation revealed small size (Wt: −5.61 SD, Ht: −3.78 SD), microcephaly (OFC: −2.75 SD), dysmorphic face, generalized muscle hypotonia, bilateral iris coloboma, and congenital cataract. Other anomalies associated: ASD, hypospadias, bilateral cryptorchidism/bilateral undescended testes and bilateral hydronephrosis. Brain MRI revealed postero-anterior interhemispheric cyst of the caudal portion of the corpus callosum. Karyotype was 46,XY and MLPA P036/P070 and P096 Probemix confirmed a ~2 Mb deletion in WHS region. Parental chromosomal analyses showed a paternal abnormal karyotype: 46,XY,1qh+,t(4;17)(p15.33;p13.3). No copy number variation was identified on 17p region by MLPA with P249 Probemix in the child.
CASE 5
Case 5 is a 1-year-old boy, the only child of an unrelated couple, with no other cases in the family. The pregnancy was uneventful, and the child was born at term (Apgar score 7, Wt-2700 g, Ht and OFC unknown). Postnatal evolution is severely delayed (can not raise head, does not speak). Last clinical examination revealed small size (Wt: −5.7 SD), microcephaly (OFC: −3.91 SD), dysmorphic face, and sacral sinus. The patient also has ASD, seizures, and frequent respiratory infections. Karyotype was 46,XY,del(4)(p16.3),del(22)(q11.23) and MLPA P245 Probemix identified a ~2 Mb deletion in WHS region in association with a small deletion in 22q11.2 region. A ~2 Mb 4p deletion was confirmed by P096 MLPA kit and the deletion in 22q11.2 in
RTDR1 gene was confirmed by P250 MLPA kit (
Figure 4).
3.2. Large Deletions (>3 Mb)
CASE 6
Case 6 is a 5-year-old girl, the first child of an unrelated young couple. The family history is negative. She presented prenatal growth delay, and she was born prematurely (33 WA; Wt = 930 g, Ht = 38 cm, OFC = 36 cm, Apgar score 1). Postnatal development was delayed (raised head at 4 Mo, sat at 8 Mo). The last evaluation (4 Y 4 Mo) revealed: proportionate dwarfism (Wt: −2.6 SD, Ht: −4.08 SD), marked microcephaly (OFC: −7.41 SD), dysmorphic face, right iris coloboma, anodontia, left preauricular pit, muscle hypotonia, severe intellectual disability, and hearing loss. The patient also presents associated congenital heart defects (ASD, VSD, tricuspid insufficiency(TI), mitral and pulmonary insufficiency. Karyotype was 46,XX,del(4)(p16.1-pter) and a ~8 Mb deletion in WHS region confirmed by FISH.
CASE 7
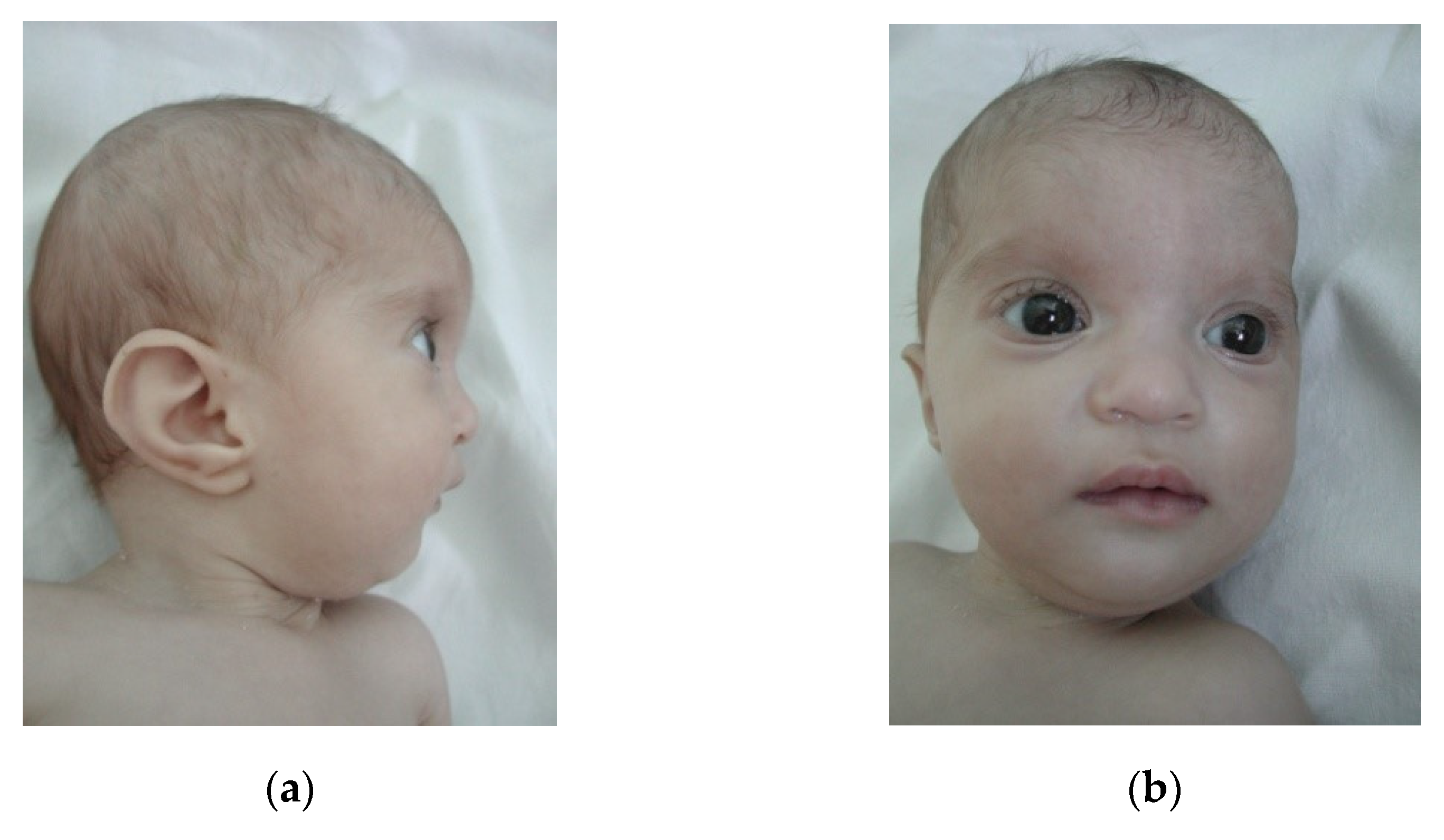
Case 7 is an 1-year-old girl, third child of a young, unrelated young couple, with no other cases in the family. Pregnancy was characterized by an imminent miscarriage (resolved by obstetrician) and an uneventful birth at term(Wt = 1600 g, Ht = 45 cm, OFC = 29 cm, Apgar score 9). The girl has a poor postnatal growth. Clinical evaluation (10 Mo) noted small size (Wt: −5.19 SD, Ht: −3.19 SD), microcephaly (OFC: −2.81 SD), characteristic dysmorphic face (
Figure 5a,b), hypotonia, developmental delay, and hearing loss (bilateral stenosis of auditory canal). Echocardiography revealed: ASD and pulmonary stenosis; recurrent respiratory infections were also noticed. Karyotype was 46,XX,del(4)(p15.2-pter) and MLPA confirmed a ~22 Mb deletion in WHS region.
Clinical features of our patients are synthetized in
Table 1.
4. Discussion
Wolf-Hirschhorn syndrome has a high clinical variability, but also a great variability of the deletion both in terms of size and etiological mechanism. Our selected cases illustrate this well. Most WHS patients had a “pure” deletion with no other cytogenetic anomalies (55%); the remaining had a more complex cytogenetic profile, such as derivative chromosome 4 resulting from an unbalanced translocation, ring 4 chromosome or a 4p-mosaicism [
10].
Cases 6 and 7 had large deletions on the short arm of chromosome 4 that was observed on the karyotype. Patient’s 6 karyotype—46,XX,del(4)(p16.1-pter)—revealed an 8.2 Mb deletion. Thus, it falls in the category of average size deletions (5 to 18 Mb). The clinical features of this specific case are similar to the ones found in a case report of a much younger girl (8 months old), both reporting impairments of the auditory system, microcephaly, coloboma, congenital heart defects, and delayed development which are considered to be some of the diagnostic markers for WHS [
11]. Case 7 is part of the third category of deletions in WHS, having a 22 Mb deletion–46,XX,del(4)(p15.2-pter). The phenotype of the two patients is more severe in terms of developmental delay, and cardiac impairment is also important. Both cases had hearing loss, aspect also illustrated by other studies in WHS cases with deletions greater than 5 MB [
12,
13]. In South et al.’s study, 13 of 32 patients have hearing loss, four of this has 4p deletions are smaller than 6 Mb, one pure deletion, and the other three show unbalanced translocations [
14].
In cases 1, 2 and 4, the syndrome was caused by microdeletions (~2 Mb) that comprise the critical region of 4p16.3, and therefore cannot be identified on the karyotype. Following the cytogenetic evaluation of the parents, a balanced translocation between chromosomes 4 and 17 was found in the father of patient 4 (46,XY,1qh+,t(4;17)(p15.33;p13.3)). This was not surprising because approximately 15% of cases with WHS are generated by a mis-segregation of derivative chromosomes in meiosis of a carrier of balanced translocation [
14]. The most common translocations in WHS are with the chromosome 8p, followed closely by translocations with 7p, 11p, 12p [
15]. Patients with an unbalanced translocation usually present some deviation from classic clinical manifestations due to modification of the phenotype by the trisomy material [
14]. In case 4, only the 2 Mb deletion in region 4p15.33→ter was important for the phenotype. However, the balanced translocation carried by the father of case 4 poses a problem in giving genetic counseling in future pregnancies. Thus, the parental couple had a 10–12% risk to have an abnormal embryo that associates either monosomy 4p15.33→ter and a small trisomy 17p13.3→ter either a small monosomy 17p13.3→ter and a trisomy 4p15.33→ter, in both situations is a high-risk pregnancy with birth of an abnormal child.
Case 3 analyses portrayed a complex chromosomal rearrangement: 46,XY,r(4)(p15.1q35)/46,XY,−4,+mar/47,XY,r(4)(p15.1q35),+mar. Using MLPA (kit P096, follow-up kit P264 and P358) only a 2.85 Mb size deletion was identified in the 4p16.3 region with no significant loss on the long arm of chromosome 4. We can presume in this case that a mechanism characterized by a telomere-to-telomere fusion which generates a pseudo-complete ring chromosome, associated with a small loss of genetic material is responsible for microdeletion in terminal part of short arm of chromosome 4. However, such cases are very rare and represent not more than >1% of WHS [
15,
16].
In case 5, MLPA test confirmed the presence of two microdeletions: one 4p16.3 and other 22q11.23. The first microdeletion was pathogenic and produced WHS. The second microdeletion involved the absence of the RTDR1 gene. The function of the protein encoded by this gene is yet unknown, but no pathogenicity has been associated with mutation in the RTDR1 gene. In order to characterize the 22q11.23 anomaly and establish the pathogenicity, it is necessary to evaluate the parents as well as complete the investigations with CMA to exclude a microdeletion that includes the RTDR1 gene (between genomic positions 20.652996-21.795392, which border the anomaly identified by MLPA P250 Probemix).
Between 90% and 100% of reported cases of WHS present epilepsy [
3], which is usually manifested in the first 3 years of life with a peak in incidence between 6 and 13 months. Fever triggered the seizure in infancy in about 70% of the 87 cases presented by Battaglia et al. [
17]. In a study of 83 patients Bi et al. proposes that the manifestations and intensity of seizures is attributed to the synergistic effect of several genes, such as
PIGG(phosphatidylinositol glycan anchor biosynthesis class G),
CPLX1(complexin-1) and
LETM1(leucine zipper and EF-hand containing transmembrane protein 1) in the WHS region; genes located within this 0.3 Mb defined as a seizure susceptibility interval (between 0.6 and 0.9 Mb from the terminus) [
12]. All of our cases cover this area of susceptibility, and the absence of seizure may be due to the young age of some patients (cases 3 and 5). Alternatively, seizures could have been very mild, and they were not noticed by caregivers.
Intrauterine growth restriction is a clinical sign present in most cases of WHS (80–90% cases) [
3,
18]. In our study, IUGR was detected in a relatively small number of cases (with deletions larger than 2 MB) but postnatal failure to thrive was a constant feature presented in all reported patients.
Heart defects are often mild in WHS, with the most common heart defect being an atrial septal defect, followed by pulmonary valve stenosis, tetralogy of Fallot, ventricular septal defect and patent ductus arteriosus (PDA). Structural heart defects occur in approximately 50% of cases [
3,
17]. Catela et al. (2009) proposed in their study the deletion of
FGFRL1(fibroblast growth factor receptor like 1) gene as a plausible candidate for the occurrence of cardiac anomalies [
13]. In our study, MLPA analysis revealed haploinsufficiency of
FGFRL1 gene in all seven cases, all but one presented mild cardiac defect. In case 6 the cardiac anomaly was more complex; the added severity can be attributed to the larger size of the chromosomal deletion.
In the case of WHS, as a contiguous gene syndrome, it is considered that the smaller the deletion is the less severe are the clinical manifestations [
7]. In our cohort study we did not identify major differences between microdeletions and large deletions. This might be explained by the existence of a critical region of the syndrome that determines most of its features [
19]. Thus,
WHSC1(Wolf-Hirschhorn syndrome candidate 1) gene, whose heterozygous deletions were observed in all seven cases, could be considered responsible for developmental delays, facial dysmorphisms, and short stature [
20]. Interestingly, in two cases presented by Zollino and her team [
21,
22], they have reported that their cases of microdeletions smaller than 2.8 Mb or none at all only showed a mild phenotype with malformations usually absent and a normal head circumference. However, in the cases 1, 2, and 4 presented by us, we have noted dysmorphic face, cardiac defects, multiple congenital anomalies and severe postnatal development which hints towards the involvement of genes other than the ones in the critical region. However, we have identified oral issues alongside psychomotor delays in case 1, 2, and 6(anodontia) similar to the findings of Limeres et al. [
23].
Concerning genetic investigations, in our experience, combined MLPA (P036/P070 or P245 followed by P279, P358, P096 or another follow up kit for 4p region) use is a reliable and rapid screening tool. The advantage of MLPA follow up kit is that they contain multiple probes for genes located in the region and in limited number of cases. Such a kit could be sufficient to establish deletion size and genes involved and this may represent an advantage compared to classical FISH. This represents an alternative strategy for cases when CMA is not easily accessible, but for the rest, CMA remains the gold standard technique for WHS diagnosis.
Even if our study did not manage to identify major differences between small size deletions and large deletions, one possible explanation for that could be reduced number of participants. Despite this, we consider that MLPA is a reliable tool for the diagnosis of WHS and, considering the cheaper and more easily accessible nature of this test, MLPA could be used as a first-tier test in the diagnosis of WHS as it is presented in this study.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}