
Congenital Fibrosis of the Extraocular Muscles: An Overview from Genetics to Management


Abstract
:1. Introduction
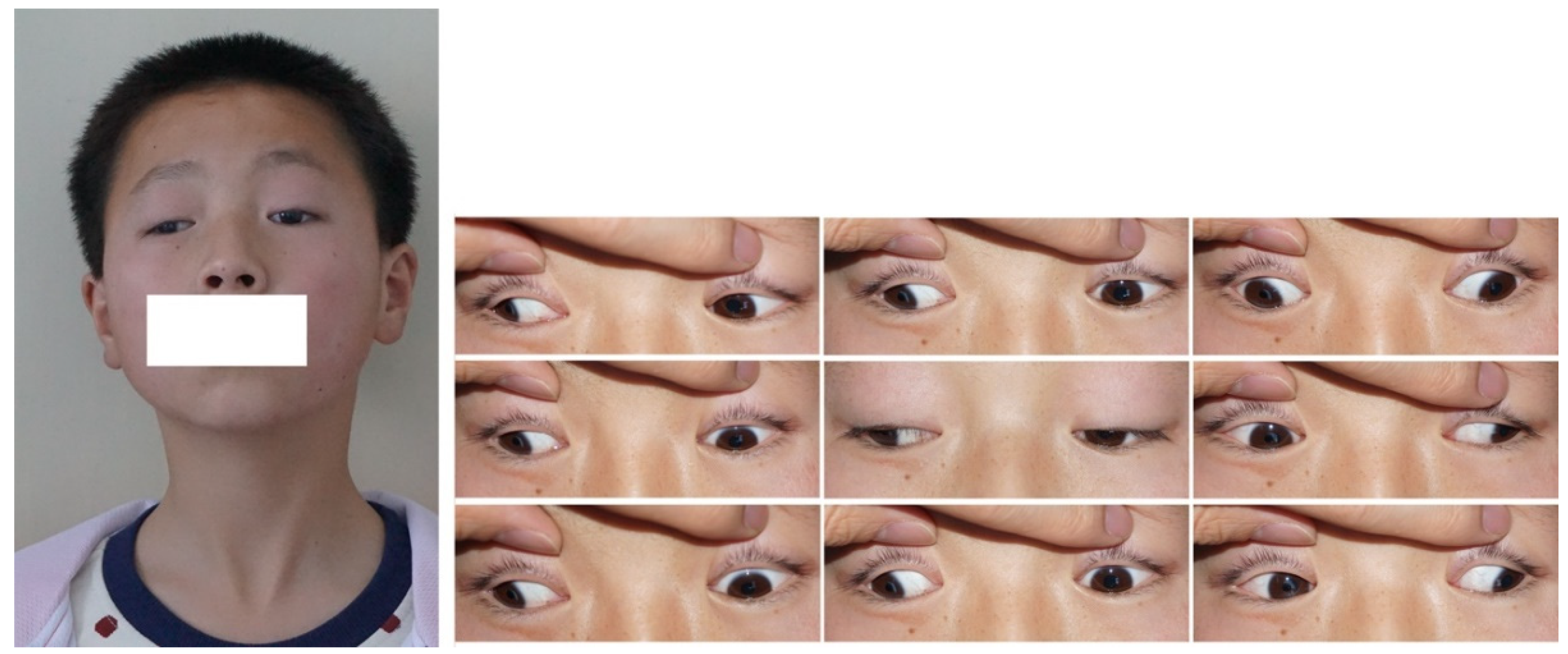
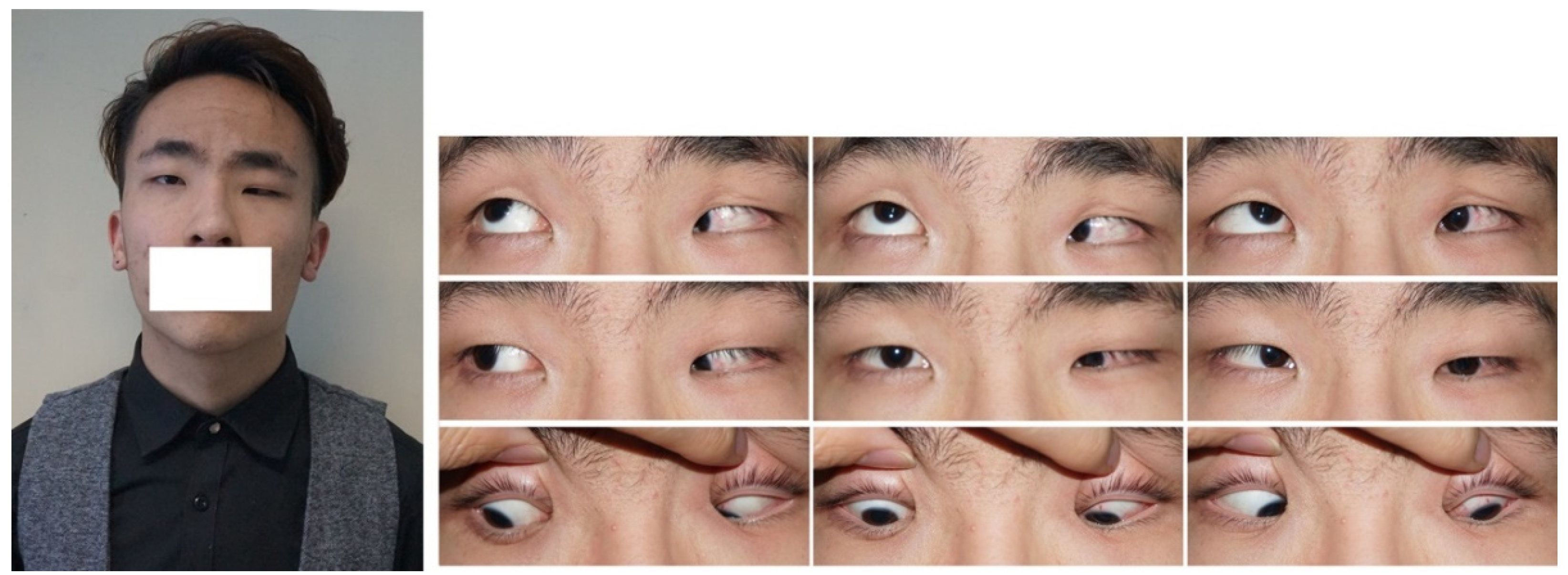
2. CFEOM Subtypes
3. Genetics
3.1. KIF21A
3.2. PHOX2A
3.3. TUBB3
3.4. TUBB2B and TUBA1A
3.5. ECEL1
3.6. COL25A1
4. Management
4.1. Initial Evaluation and Routine Monitoring
4.2. Surgical Management
4.3. Nonsurgical Management
5. Prospects: Optimizing Decisions upon Molecular Genetic Diagnoses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heidary, G.; Engle, E.C.; Hunter, D.G. Congenital fibrosis of the extraocular muscles. Semin. Ophthalmol. 2008, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Reck, A.C.; Manners, R.; Hatchwell, E. Phenotypic heterogeneity may occur in congenital fibrosis of the extraocular muscles. Br. J. Ophthalmol. 1998, 82, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivian, A.J. Congenital fibrosis of the extra-ocular muscles (CFEOM) and the cranial dysinnervation disorders. Eye 2020, 34, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Maconachie, G.D.E.; Kuht, H.J.; Chan, W.M.; Sheth, V.; Hisaund, M.; McLean, R.J.; Barry, B.; Al-Diri, B.; Proudlock, F.A.; et al. Optic Nerve Head and Retinal Abnormalities Associated with Congenital Fibrosis of the Extraocular Muscles. Int. J. Mol. Sci. 2021, 22, 2575. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Almutlaq, M.; Oystreck, D.T.; Engle, E.C.; Abu-Amero, K.; Bosley, T. Retinal Dysfunction in Patients with Congenital Fibrosis of the Extraocular Muscles Type 2. Ophthalmic Genet. 2016, 37, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.K.; Kim, J.H.; Hwang, J.M. Absent trochlear and abducens nerves in a patient with congenital fibrosis of extraocular muscles. Neurol. Sci. 2020, 41, 977–979. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, J.M. Hypoplastic oculomotor nerve and absent abducens nerve in congenital fibrosis syndrome and synergistic divergence with magnetic resonance imaging. Ophthalmology 2005, 112, 728–732. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X.; Sun, X.; Liu, H.; Zhao, K.; Chang, Q.; Pan, X.; Wang, X.; Yuan, S.; Liu, Q.; et al. Maternal germline mosaicism of kinesin family member 21A (KIF21A) mutation causes complex phenotypes in a Chinese family with congenital fibrosis of the extraocular muscles. Mol. Vis. 2014, 20, 15–23. [Google Scholar]
- Yazdani, A.; Chung, D.C.; Abbaszadegan, M.R.; Al-Khayer, K.; Chan, W.M.; Yazdani, M.; Ghodsi, K.; Engle, E.C.; Traboulsi, E.I. A novel PHOX2A/ARIX mutation in an Iranian family with congenital fibrosis of extraocular muscles type 2 (CFEOM2). Am. J. Ophthalmol. 2003, 136, 861–865. [Google Scholar] [CrossRef]
- Razek, A.; Maher, H.; Kasem, M.A.; Helmy, E. Imaging of congenital cranial dysinnervation disorders: What radiologist wants to know? Clin. Imaging 2021, 71, 106–116. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Lee, J.H.; Park, H.J.; Choi, Y.R.; Hyun, Y.S.; Park, J.H.; Koo, H.; Chung, K.W.; Choi, B.O. A family with axonal sensorimotor polyneuropathy with TUBB3 mutation. Mol. Med. Rep. 2015, 11, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.G.; Maconachie, G.D.E.; Constantinescu, C.S.; Chan, W.M.; Barry, B.; Hisaund, M.; Sheth, V.; Kuht, H.J.; Dineen, R.A.; Harieaswar, S.; et al. Congenital monocular elevation deficiency associated with a novel TUBB3 gene variant. Br. J. Ophthalmol. 2020, 104, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Ma, J.; Zhang, T.; Ma, D. Advances in the Genetics of Congenital Ptosis. Ophthalmic Res. 2022, 65, 131–139. [Google Scholar] [CrossRef]
- Tukel, T.; Uzumcu, A.; Gezer, A.; Kayserili, H.; Yuksel-Apak, M.; Uyguner, O.; Gultekin, S.H.; Hennies, H.C.; Nurnberg, P.; Desnick, R.J.; et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. J. Med. Genet. 2005, 42, 408–415. [Google Scholar] [CrossRef]
- Munezane, H.; Oizumi, H.; Wakabayashi, T.; Nishio, S.; Hirasawa, T.; Sato, T.; Harada, A.; Yoshida, T.; Eguchi, T.; Yamanashi, Y.; et al. Roles of Collagen XXV and Its Putative Receptors PTPσ/δ in Intramuscular Motor Innervation and Congenital Cranial Dysinnervation Disorder. Cell Rep. 2019, 29, 4362–4376.e4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.C.; Jurgens, J.A.; Hunter, D.G.; Engle, E.C. Congenital Fibrosis of the Extraocular Muscles Overview. In GeneReviews(R); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chen, X.; Guo, X.; Ma, H.Z. A clinical analysis of 40 cases with congenital fibrosis of extraocular muscles. Zhonghua Yan Ke Za Zhi 2011, 47, 978–982. [Google Scholar] [PubMed]
- Yazdani, A.; Traboulsi, E.I. Classification and surgical management of patients with familial and sporadic forms of congenital fibrosis of the extraocular muscles. Ophthalmology 2004, 111, 1035–1042. [Google Scholar] [CrossRef]
- Wang, S.M.; Zwaan, J.; Mullaney, P.B.; Jabak, M.H.; Al-Awad, A.; Beggs, A.H.; Engle, E.C. Congenital fibrosis of the extraocular muscles type 2, an inherited exotropic strabismus fixus, maps to distal 11q13. Am. J. Hum. Genet. 1998, 63, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Shoshany, T.N.; Robson, C.D.; Hunter, D.G. Anomalous superior oblique muscles and tendons in congenital fibrosis of the extraocular muscles. J. AAPOS 2019, 23, 325.e1–325.e6. [Google Scholar] [CrossRef]
- Engle, E.C.; McIntosh, N.; Yamada, K.; Lee, B.A.; Johnson, R.; O’Keefe, M.; Letson, R.; London, A.; Ballard, E.; Ruttum, M.; et al. CFEOM1, the classic familial form of congenital fibrosis of the extraocular muscles, is genetically heterogeneous but does not result from mutations in ARIX. BMC Genet. 2002, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Sener, E.C.; Taylan Sekeroglu, H.; Ural, O.; Oztürk, B.T.; Sanaç, A.S. Strabismus surgery in congenital fibrosis of the extraocular muscles: A paradigm. Ophthalmic Genet. 2014, 35, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Price, J.M.; Boparai, R.S.; Wasserman, B.N. Congenital fibrosis of the extraocular muscles: Review of recent literature. Curr. Opin. Ophthalmol. 2019, 30, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Khalil, D.S.; Al Sharif, L.J.; Al-Ghadhfan, F.E.; Al Tassan, N.A. Germline Mosaicism for KIF21A Mutation (p.R954L) Mimicking Recessive Inheritance for Congenital Fibrosis of the Extraocular Muscles. Ophthalmology 2010, 117, 154–158. [Google Scholar] [CrossRef]
- Di Fabio, R.; Comanducci, G.; Piccolo, F.; Santorelli, F.M.; De Berardinis, T.; Tessa, A.; Sabatini, U.; Pierelli, F.; Casali, C. Cerebellar atrophy in congenital fibrosis of the extraocular muscles type 1. Cerebellum 2013, 12, 140–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliani, L.; Spagnoli, C.; Salerno, G.G.; Mehine, M.; Rizzi, S.; Frattini, D.; Koskenvuo, J.; Fusco, C. A Novel De Novo KIF21A Variant in a Patient With Congenital Fibrosis of the Extraocular Muscles With a Syndromic CFEOM Phenotype. J. Neuroophthalmol. 2021, 41, e85–e88. [Google Scholar] [CrossRef]
- Xia, C.R.; Shi, L.H.; Nan, J.; Hao, Y.Z.; Jia, Y. Identification of a novel KIF21A gene mutation in a Chinese family with congenital fibrosis of the extraocular muscles. Zhonghua Yan Ke Za Zhi 2022, 58, 213–214. [Google Scholar] [CrossRef]
- Ali, Z.; Xing, C.; Anwar, D.; Itani, K.; Weakley, D.; Gong, X.; Pascual, J.M.; Mootha, V.V. A novel de novo KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Möbius syndrome. Mol. Vis. 2014, 20, 368–375. [Google Scholar]
- Yamada, K.; Chan, W.M.; Andrews, C.; Bosley, T.M.; Sener, E.C.; Zwaan, J.T.; Mullaney, P.B.; Oztürk, B.T.; Akarsu, A.N.; Sabol, L.J.; et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Invest. Ophthalmol. Vis. Sci. 2004, 45, 2218–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Zhao, C.; Zhao, K.; Li, N.; Larsson, C. Novel and recurrent KIF21A mutations in congenital fibrosis of the extraocular muscles type 1 and 3. Arch. Ophthalmol. 2008, 126, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yamada, K.; Katz, B.; Guan, H.; Wang, L.; Andrews, C.; Zhao, G.; Engle, E.C.; Chen, H.; Tong, Z.; et al. KIF21A mutations in two Chinese families with congenital fibrosis of the extraocular muscles (CFEOM). Mol. Vis. 2010, 16, 2062–2070. [Google Scholar] [PubMed]
- Yamada, K.; Hunter, D.G.; Andrews, C.; Engle, E.C. A novel KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn jaw-winking phenomenon. Arch. Ophthalmol. 2005, 123, 1254–1259. [Google Scholar] [CrossRef]
- Wang, P.; Li, S.; Xiao, X.; Guo, X.; Zhang, Q. KIF21A novel deletion and recurrent mutation in patients with congenital fibrosis of the extraocular muscles-1. Int. J. Mol. Med. 2011, 28, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Al-Haddad, C.; Boustany, R.M.; Rachid, E.; Ismail, K.; Barry, B.; Chan, W.M.; Engle, E. KIF21A pathogenic variants cause congenital fibrosis of extraocular muscles type 3. Ophthalmic Genet. 2021, 42, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; Andrews, C.; Dragan, L.; Fredrick, D.; Armstrong, L.; Lyons, C.; Geraghty, M.T.; Hunter, D.G.; Yazdani, A.; Traboulsi, E.I.; et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet. 2007, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed] [Green Version]
- van der Vaart, B.; van Riel, W.E.; Doodhi, H.; Kevenaar, J.T.; Katrukha, E.A.; Gumy, L.; Bouchet, B.P.; Grigoriev, I.; Spangler, S.A.; Yu, K.L.; et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev. Cell 2013, 27, 145–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Andrews, C.; Chan, W.M.; McKeown, C.A.; Magli, A.; de Berardinis, T.; Loewenstein, A.; Lazar, M.; O’Keefe, M.; Letson, R.; et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat. Genet. 2003, 35, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Desai, J.; Miranda, C.J.; Duncan, J.S.; Qiu, W.; Nugent, A.A.; Kolpak, A.L.; Wu, C.C.; Drokhlyansky, E.; Delisle, M.M.; et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 2014, 82, 334–349. [Google Scholar] [CrossRef] [Green Version]
- Bosley, T.M.; Oystreck, D.T.; Robertson, R.L.; al Awad, A.; Abu-Amero, K.; Engle, E.C. Neurological features of congenital fibrosis of the extraocular muscles type 2 with mutations in PHOX2A. Brain 2006, 129, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.M.; Shen, Q.; Li, J.; Du, W.; Pang, H.L.; Lin, S.F.; Bu, J. Identification of a novel PHOX2A gene mutation in a Chinese family with congenital fibrosis of extraocular muscles type 2. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2012, 29, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Yamada, K.; Fain, J.; Sener, E.C.; Selleck, C.J.; Awad, A.H.; Zwaan, J.; Mullaney, P.B.; Bosley, T.M.; Engle, E.C. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat. Genet. 2001, 29, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Matsuo, T.; Fujiwara, H.; Hasebe, S.; Ohtsuki, H.; Yasuda, T. ARIX gene polymorphisms in patients with congenital superior oblique muscle palsy. Br. J. Ophthalmol. 2004, 88, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkubo, S.I.; Matsuo, T.; Hasebe, K.; Shira, Y.H.; Itoshima, E.; Ohtsuki, H. Phenotype-phenotype and genotype-phenotype correlations in patients with idiopathic superior oblique muscle palsy. J. Hum. Genet. 2012, 57, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latremoliere, A.; Cheng, L.; DeLisle, M.; Wu, C.; Chew, S.; Hutchinson, E.B.; Sheridan, A.; Alexandre, C.; Latremoliere, F.; Sheu, S.H.; et al. Neuronal-Specific TUBB3 Is Not Required for Normal Neuronal Function but Is Essential for Timely Axon Regeneration. Cell Rep. 2018, 24, 1865–1879.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.C.; Engle, E.C. Ocular congenital cranial dysinnervation disorders (CCDDs): Insights into axon growth and guidance. Hum. Mol. Genet. 2017, 26, R37–R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demer, J.L.; Clark, R.A.; Tischfield, M.A.; Engle, E.C. Evidence of an asymmetrical endophenotype in congenital fibrosis of extraocular muscles type 3 resulting from TUBB3 mutations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4600–4611. [Google Scholar] [CrossRef] [Green Version]
- Whitman, M.C.; Andrews, C.; Chan, W.M.; Tischfield, M.A.; Stasheff, S.F.; Brancati, F.; Ortiz-Gonzalez, X.; Nuovo, S.; Garaci, F.; MacKinnon, S.E.; et al. Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am. J. Med. Genet. A 2016, 170, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.C.; Barry, B.J.; Robson, C.D.; Facio, F.M.; Van Ryzin, C.; Chan, W.M.; Lehky, T.J.; Thurm, A.; Zalewski, C.; King, K.A.; et al. TUBB3 Arg262His causes a recognizable syndrome including CFEOM3, facial palsy, joint contractures, and early-onset peripheral neuropathy. Hum. Genet. 2021, 140, 1709–1731. [Google Scholar] [CrossRef]
- Jang, Y.; Kwak, E.; An, J.Y.; Jung, J.H. Infantile esotropia in a family with TUBB3 mutation associated congenital fibrosis of extraocular muscles. Ophthalmic Genet. 2022, 43, 716–719. [Google Scholar] [CrossRef]
- Xue, J.; Song, Z.; Ma, S.; Yi, Z.; Yang, C.; Li, F.; Liu, K.; Zhang, Y. A Novel De Novo TUBB3 Variant Causing Developmental Delay, Epilepsy and Mild Ophthalmological Symptoms in a Chinese Child. J. Mol. Neurosci. 2022, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, I.; et al. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Olney, A.H.; Beavers, A.; Spaulding, J.; Nelson, M.; Nielsen, S.; Sanmann, J.N. The recurrent TUBB3 Gly98Ser substitution is the first described to inconsistently result in CFEOM3. Am. J. Med. Genet. A 2020, 182, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Park, S.E.; Won, D.; Lee, S.T.; Han, S.H.; Han, J. TUBB3 M323V Syndrome Presents with Infantile Nystagmus. Genes 2021, 12, 575. [Google Scholar] [CrossRef] [PubMed]
- Dentici, M.L.; Maglione, V.; Agolini, E.; Catena, G.; Capolino, R.; Lanari, V.; Novelli, A.; Sinibaldi, L.; Vecchio, D.; Gonfiantini, M.V.; et al. TUBB3 E410K syndrome: Case report and review of the clinical spectrum of TUBB3 mutations. Am. J. Med. Genet. A 2020, 182, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Cederquist, G.Y.; Luchniak, A.; Tischfield, M.A.; Peeva, M.; Song, Y.; Menezes, M.P.; Chan, W.M.; Andrews, C.; Chew, S.; Jamieson, R.V.; et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum. Mol. Genet. 2012, 21, 5484–5499. [Google Scholar] [CrossRef] [Green Version]
- Jurgens, J.A.; Barry, B.J.; Lemire, G.; Chan, W.M.; Whitman, M.C.; Shaaban, S.; Robson, C.D.; MacKinnon, S.; England, E.M.; McMillan, H.J.; et al. Novel variants in TUBA1A cause congenital fibrosis of the extraocular muscles with or without malformations of cortical brain development. Eur. J. Hum. Genet. 2021, 29, 816–826. [Google Scholar] [CrossRef]
- Romaniello, R.; Arrigoni, F.; Panzeri, E.; Poretti, A.; Micalizzi, A.; Citterio, A.; Bedeschi, M.F.; Berardinelli, A.; Cusmai, R.; D’Arrigo, S.; et al. Tubulin-related cerebellar dysplasia: Definition of a distinct pattern of cerebellar malformation. Eur. Radiol. 2017, 27, 5080–5092. [Google Scholar] [CrossRef]
- Ullmann, U.; D’Argenzio, L.; Mathur, S.; Whyte, T.; Quinlivan, R.; Longman, C.; Farrugia, M.E.; Manzur, A.; Willis, T.; Jungbluth, H.; et al. ECEL1 gene related contractural syndrome: Long-term follow-up and update on clinical and pathological aspects. Neuromuscul. Disord. 2018, 28, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Huddar, A.; Polavarapu, K.; Preethish-Kumar, V.; Bardhan, M.; Unnikrishnan, G.; Nashi, S.; Vengalil, S.; Priyadarshini, P.; Kulanthaivelu, K.; Arunachal, G.; et al. Expanding the Phenotypic Spectrum of ECEL1-Associated Distal Arthrogryposis. Children 2021, 8, 909. [Google Scholar] [CrossRef]
- Shaaban, S.; Duzcan, F.; Yildirim, C.; Chan, W.M.; Andrews, C.; Akarsu, N.A.; Engle, E.C. Expanding the phenotypic spectrum of ECEL1-related congenital contracture syndromes. Clin. Genet. 2014, 85, 562–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.O.; Shaheen, R.; Alkuraya, F.S. The ECEL1-related strabismus phenotype is consistent with congenital cranial dysinnervation disorder. J. AAPOS 2014, 18, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, K.; Quijano-Roy, S.; Monnier, N.; Zhou, J.; Fauré, J.; Smirnow, D.A.; Carlier, R.; Laroche, C.; Marcorelles, P.; Mercier, S.; et al. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum. Mol. Genet. 2013, 22, 1483–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.O.; Al-Mesfer, S. Recessive COL25A1 mutations cause isolated congenital ptosis or exotropic Duane syndrome with synergistic divergence. J. AAPOS 2015, 19, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Shinwari, J.M.; Khan, A.; Awad, S.; Shinwari, Z.; Alaiya, A.; Alanazi, M.; Tahir, A.; Poizat, C.; Al Tassan, N. Recessive mutations in COL25A1 are a cause of congenital cranial dysinnervation disorder. Am. J. Hum. Genet. 2015, 96, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natera-de Benito, D.; Jurgens, J.A.; Yeung, A.; Zaharieva, I.T.; Manzur, A.; DiTroia, S.P.; Di Gioia, S.A.; Pais, L.; Pini, V.; Barry, B.J.; et al. Recessive variants in COL25A1 gene as novel cause of arthrogryposis multiplex congenita with ocular congenital cranial dysinnervation disorder. Hum. Mutat. 2022, 43, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Oystreck, D.T. Ophthalmoplegia and Congenital Cranial Dysinnervation Disorders. J. Binocul. Vis. Ocul. Motil. 2018, 68, 31–33. [Google Scholar] [CrossRef]
- Khan, A.O. Outcomes of strabismus surgery in genetically confirmed congenital fibrosis of the extraocular muscles. J. AAPOS 2020, 24, 127–128. [Google Scholar] [CrossRef]
- Shaaban, S.; Ramos-Platt, L.; Gilles, F.H.; Chan, W.M.; Andrews, C.; De Girolami, U.; Demer, J.; Engle, E.C. RYR1 mutations as a cause of ophthalmoplegia, facial weakness, and malignant hyperthermia. JAMA Ophthalmol. 2013, 131, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Lawal, T.A.; Todd, J.J.; Witherspoon, J.W.; Bönnemann, C.G.; Dowling, J.J.; Hamilton, S.L.; Meilleur, K.G.; Dirksen, R.T. Ryanodine receptor 1-related disorders: An historical perspective and proposal for a unified nomenclature. Skelet. Muscle 2020, 10, 32. [Google Scholar] [CrossRef]
- Heidary, G.; Mackinnon, S.; Elliott, A.; Barry, B.J.; Engle, E.C.; Hunter, D.G. Outcomes of strabismus surgery in genetically confirmed congenital fibrosis of the extraocular muscles. J. AAPOS 2019, 23, 253.e1–253.e6. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.; Balasubramanian, R.; Chan, W.M.; Kang, P.B.; Andrews, C.; Webb, B.D.; MacKinnon, S.E.; Oystreck, D.T.; Rankin, J.; Crawford, T.O.; et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain 2013, 136, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Sharma, P.; Gaur, N.; Singh, J. Selective aplasia of global fibres of all extraocular muscles in congenital fibrosis of extraocular muscles (CFEOM): A rare presentation. BMJ Case Rep. 2017, 2017, bcr-2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apt, L.; Axelrod, R.N. Generalized fibrosis of the extraocular muscles. Am. J. Ophthalmol. 1978, 85, 822–829. [Google Scholar] [CrossRef]
- Tawfik, H.A.; Rashad, M.A. Surgically mismanaged ptosis in a patient with congenital fibrosis of the extraocular muscles type I. Middle East Afr. J. Ophthalmol. 2012, 19, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Hedergott, A.; Pink-Theofylaktopoulos, U.; Neugebauer, A.; Fricke, J. Tendon elongation with bovine pericardium in strabismus surgery-indications beyond Graves’ orbitopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2021, 259, 145–155. [Google Scholar] [CrossRef]
- Cestari, D.M.; Hunter, D.G. Learning Strabismus Surgery: A Case-Based Approach; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 192–197. [Google Scholar]
- Al-Haddad, C.E.; Abdulaal, M. Transposition surgery for inferior rectus fibrosis. J. Pediatr. Ophthalmol. Strabismus 2015, 52, e1–e3. [Google Scholar] [CrossRef]
- Magli, A.; de Berardinis, T.; D’Esposito, F.; Gagliardi, V. Clinical and surgical data of affected members of a classic CFEOM I family. BMC Ophthalmol. 2003, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.R.; Baek, S.H.; Kim, U.S. Dissociated vertical deviation in congenital fibrosis of the extraocular muscles. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 1007–1008. [Google Scholar] [CrossRef]
- Letson, R.D. Surgical Management of the Ocular Congenital Fibrosis Syndrome. Am. Orthopt. J. 1980, 30, 97–100. [Google Scholar] [CrossRef]
- Okita, Y.; Kimura, A.; Okamoto, M.; Mimura, O.; Gomi, F. Surgical management of pediatric patients with congenital fibrosis of the extraocular muscles. Jpn. J. Ophthalmol. 2020, 64, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, H.A.; Rashad, M.A. Surgical management of hypotropia in congenital fibrosis of extraocular muscles (CFEOM) presented by pseudoptosis. Clin. Ophthalmol. 2013, 7, 1–6. [Google Scholar] [CrossRef]
- Doherty, E.J.; Macy, M.E.; Wang, S.M.; Dykeman, C.P.; Melanson, M.T.; Engle, E.C. CFEOM3: A new extraocular congenital fibrosis syndrome that maps to 16q24.2-q24.3. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1687–1694. [Google Scholar]
- Heidary, G.; Hunter, D.G. Reply. J. AAPOS 2020, 24, 128. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, H.J.; Kim, S.J. Clinical Features of Duane Retraction Syndrome: A New Classification. Korean J. Ophthalmol. 2020, 34, 158–165. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Locus | Inheritance | Penetrance | Phenotype | Featured Genotype–Phenotype Correlations |
|---|---|---|---|---|
| KIF21A | Autosomal dominant | Complete | CFEOM-1 (common) or CFEOM-3 (rare) | Large-angle exo-hypotropia Marcus Gunn jaw winking |
| PHOX2A | Autosomal recessive | Complete | CFEOM-2 | Large-angle exotropia Pupil anomalies |
| TUBB3 | Autosomal dominant | Incomplete | CFEOM-1 (rare) or CFEOM-3 (common) | Malformations of cortical development Cognitive impairment Significant phenotypic variability E410K syndrome R262H syndrome |
| TUBB2B | Autosomal dominant | Complete | CFEOM-3 | Polymicrogyria Cognitive impairment |
| TUBA1A | Autosomal dominant | Unknown | CFEOM-1 or CFEOM-3 | Polymicrogyria Cognitive impairment |
| ECEL1 | Autosomal recessive | Incomplete | Undefinable | Arthrogryposis multiplex congenita |
| COL25A1 | Autosomal recessive | Unknown | CFEOM-5 | Exotropia Globe retraction with synergistic divergence Arthrogryposis multiplex congenita |
| Locus on chromosome 21qter | Autosomal recessive | Unknown | CFEOM-4 (Tukel syndrome) | Hand oligodactyly |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, W.; Wei, Y.; Wu, L.; Zhao, C. Congenital Fibrosis of the Extraocular Muscles: An Overview from Genetics to Management. Children 2022, 9, 1605. https://doi.org/10.3390/children9111605
Xia W, Wei Y, Wu L, Zhao C. Congenital Fibrosis of the Extraocular Muscles: An Overview from Genetics to Management. Children. 2022; 9(11):1605. https://doi.org/10.3390/children9111605
Chicago/Turabian StyleXia, Weiyi, Yan Wei, Lianqun Wu, and Chen Zhao. 2022. "Congenital Fibrosis of the Extraocular Muscles: An Overview from Genetics to Management" Children 9, no. 11: 1605. https://doi.org/10.3390/children9111605
APA StyleXia, W., Wei, Y., Wu, L., & Zhao, C. (2022). Congenital Fibrosis of the Extraocular Muscles: An Overview from Genetics to Management. Children, 9(11), 1605. https://doi.org/10.3390/children9111605