
Atypical Presentation of Enlarged Vestibular Aqueducts Caused by SLC26A4 Variants


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
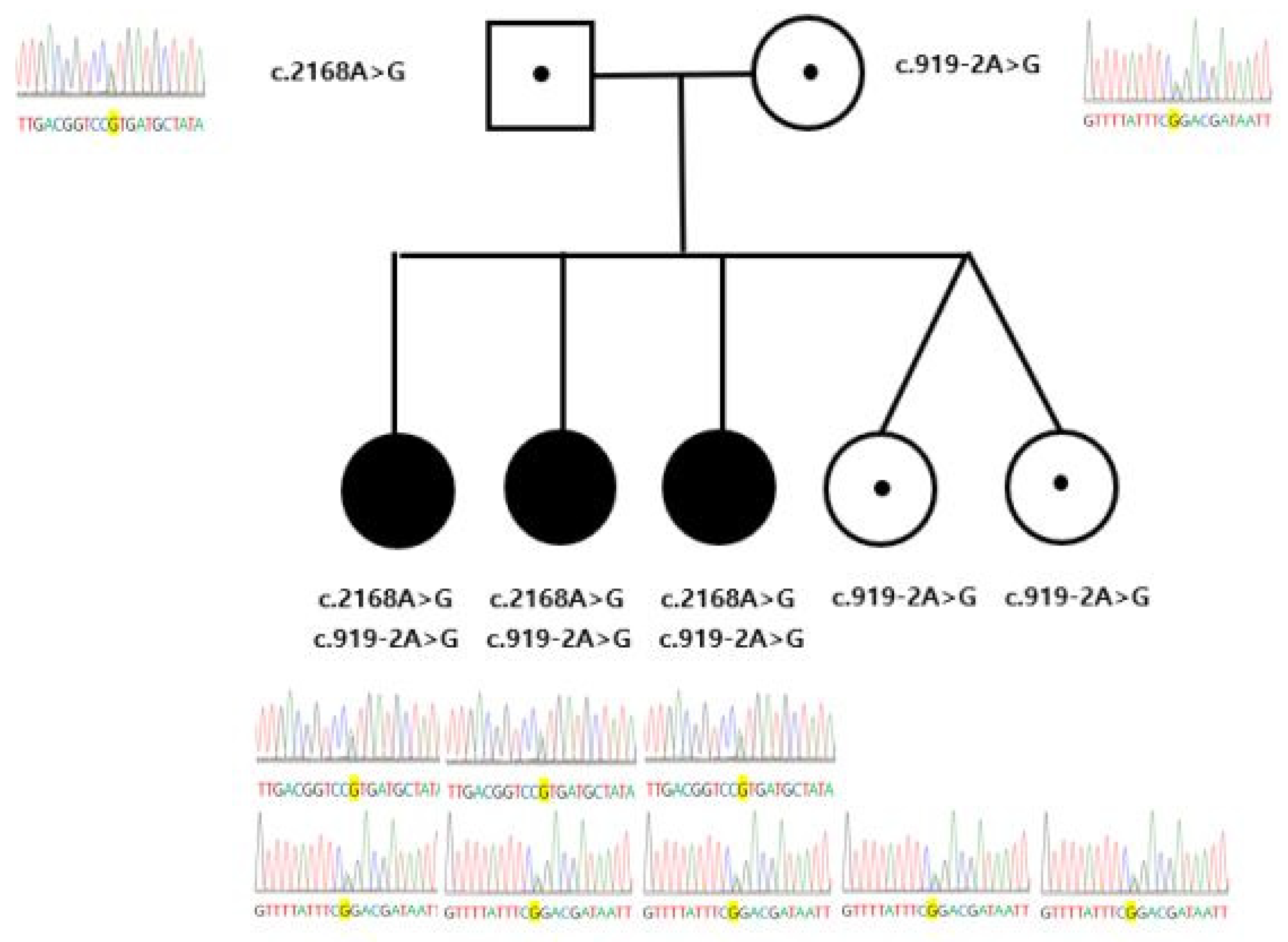
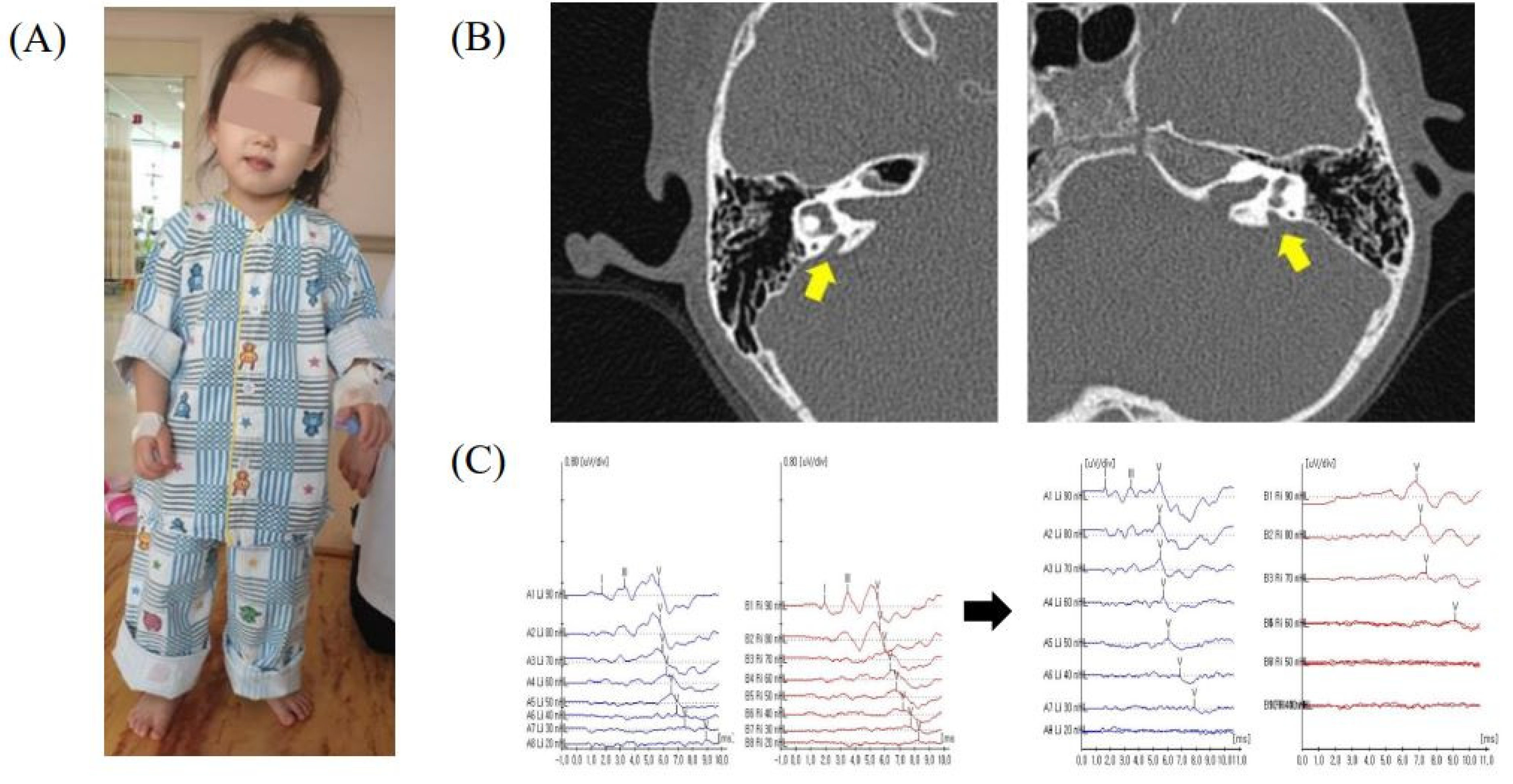
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fortnum, H.M.; Summerfield, A.Q.; Marshall, D.H.; Davis, A.C.; Bamford, J.M. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: Questionnaire based ascertainment study. BMJ 2001, 323, 536–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olusanya, B.O.; Davis, A.C.; Hoffman, H.J. Hearing loss: Rising prevalence and impact. Bull. World Health Organ. 2019, 97, 646–646A. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Li, D.; Wang, P.; Fan, D.; De, J.; Zhu, W. Autosomal recessive non-syndromic hearing loss is caused by novel compound heterozygous mutations in TMC1 from a Tibetan Chinese family. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Svidnicki, M.C.; Silva-Costa, S.M.; Ramos, P.Z.; dos Santos, N.Z.; Martins, F.T.; Castilho, A.M.; Sartorato, E.L. Screening of genetic alterations related to non-syndromic hearing loss using MassARRAY iPLEX(R) technology. BMC Med. Genet. 2015, 16, 85. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.; Pang, X.; Chen, D.; Li, L.; Chen, Y.; Sun, L.; Wang, X.; Wu, H.; Yang, T. Molecular etiology of non-dominant, non-syndromic, mild-to-moderate childhood hearing impairment in Chinese Hans. Am. J. Med. Genet. A 2014, 164A, 3115–3119. [Google Scholar] [CrossRef]
- De Moraes, V.C.S.; Bernardinelli, E.; Zocal, N.; Fernandez, J.A.; Nofziger, C.; Castilho, A.M.; Sartorato, E.L.; Paulmichl, M.; Dossena, S. Reduction of Cellular Expression Levels Is a Common Feature of Functionally Affected Pendrin (SLC26A4) Protein Variants. Mol. Med. 2016, 22, 41–53. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Mila, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef] [Green Version]
- Miyagawa, M.; Nishio, S.Y.; Usami, S.; Deafness Gene Study, C. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.J.; Zhao, Y.L.; Rao, S.Q.; Guo, Y.F.; Yuan, H.; Zong, L.; Guan, J.; Xu, B.C.; Wang, D.Y.; Han, M.K.; et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 2007, 72, 245–254. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.J.; Jin, H.S.; Lee, J.O.; Go, S.H.; Jang, H.S.; Moon, S.K.; Lee, S.C.; Chun, Y.M.; Lee, H.K.; et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 2005, 67, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Wemeau, J.L.; Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, J.; Efrati, E.; Adler, L.; Tal, O.; Carrithers, S.L.; Alper, S.L.; Zelikovic, I. Transcriptional regulation of the pendrin gene. Cell. Physiol. Biochem. 2011, 28, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, J.; Tal, O.; Kladnitsky, O.; Adler, L.; Efrati, E.; Carrithers, S.L.; Alper, S.L.; Zelikovic, I. The pendrin anion exchanger gene is transcriptionally regulated by uroguanylin: A novel enterorenal link. Am. J. Physiol. Renal Physiol. 2012, 302, F614–F624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, N.; Fugazzola, L.; Evans, M.; Chatterjee, K.; Karet, F. Life-threatening metabolic alkalosis in Pendred syndrome. Eur. J. Endocrinol. 2011, 165, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Nakao, I.; Kanaji, S.; Ohta, S.; Matsushita, H.; Arima, K.; Yuyama, N.; Yamaya, M.; Nakayama, K.; Kubo, H.; Watanabe, M.; et al. Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J. Immunol. 2008, 180, 6262–6269. [Google Scholar] [CrossRef] [Green Version]
- Hone, S.W.; Smith, R.J. Genetic screening for hearing loss. Clin. Otolaryngol. Allied Sci. 2003, 28, 285–290. [Google Scholar] [CrossRef]
- Boston, M.; Halsted, M.; Meinzen-Derr, J.; Bean, J.; Vijayasekaran, S.; Arjmand, E.; Choo, D.; Benton, C.; Greinwald, J. The large vestibular aqueduct: A new definition based on audiologic and computed tomography correlation. Otolaryngol. Head Neck Surg. 2007, 136, 972–977. [Google Scholar] [CrossRef]
- Steinbach, S.; Brockmeier, S.J.; Kiefer, J. The large vestibular aqueduct--case report and review of the literature. Acta Otolaryngol. 2006, 126, 788–795. [Google Scholar] [CrossRef]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef]
- Coyle, B.; Reardon, W.; Herbrick, J.A.; Tsui, L.C.; Gausden, E.; Lee, J.; Coffey, R.; Grueters, A.; Grossman, A.; Phelps, P.D.; et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum. Mol. Genet. 1998, 7, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, S.; Riazuddin, S.; Ahmed, Z.M.; Tasneem, S.; Jaleel, A.; Khan, S.Y.; Griffith, A.J.; Friedman, T.B.; Riazuddin, S. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J. Hum. Genet. 2009, 54, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Stewart, A.K.; Madeo, A.C.; Pryor, S.P.; Lenhard, S.; Kittles, R.; Eisenman, D.; Kim, H.J.; Niparko, J.; Thomsen, J.; et al. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: Genotype-phenotype correlation or coincidental polymorphisms? Hum. Mutat. 2009, 30, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Yeh, T.H.; Chen, P.J.; Hsu, C.J. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: A unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope 2005, 115, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Jackler, R.K.; De La Cruz, A. The large vestibular aqueduct syndrome. Laryngoscope 1989, 99, 1238–1242. [Google Scholar] [CrossRef]
- Levenson, M.J.; Parisier, S.C.; Jacobs, M.; Edelstein, D.R. The large vestibular aqueduct syndrome in children. A review of 12 cases and the description of a new clinical entity. Arch. Otolaryngol. Head Neck Surg. 1989, 115, 54–58. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, K.; Zhang, X.; Peng, K.A. Fluctuating Sensorineural Hearing Loss. Audiol. Neurootol. 2019, 24, 109–116. [Google Scholar] [CrossRef]
- Noordman, B.J.; van Beeck Calkoen, E.; Witte, B.; Goverts, T.; Hensen, E.; Merkus, P. Prognostic factors for sudden drops in hearing level after minor head injury in patients with an enlarged vestibular aqueduct: A meta-analysis. Otol. Neurotol. 2015, 36, 4–11. [Google Scholar] [CrossRef]
- Kanagalingam, S.; Miller, N.R. Horner syndrome: Clinical perspectives. Eye Brain 2015, 7, 35–46. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byun, J.C.; Lee, K.-Y.; Hwang, S.-K. Atypical Presentation of Enlarged Vestibular Aqueducts Caused by SLC26A4 Variants. Children 2022, 9, 165. https://doi.org/10.3390/children9020165
Byun JC, Lee K-Y, Hwang S-K. Atypical Presentation of Enlarged Vestibular Aqueducts Caused by SLC26A4 Variants. Children. 2022; 9(2):165. https://doi.org/10.3390/children9020165
Chicago/Turabian StyleByun, Jun Chul, Kyu-Yup Lee, and Su-Kyeong Hwang. 2022. "Atypical Presentation of Enlarged Vestibular Aqueducts Caused by SLC26A4 Variants" Children 9, no. 2: 165. https://doi.org/10.3390/children9020165