
NRXN1 Deletion in Two Twins’ Genotype and Phenotype: A Clinical Case and Literature Review

 ,
,
Abstract
:1. Introduction
2. Report
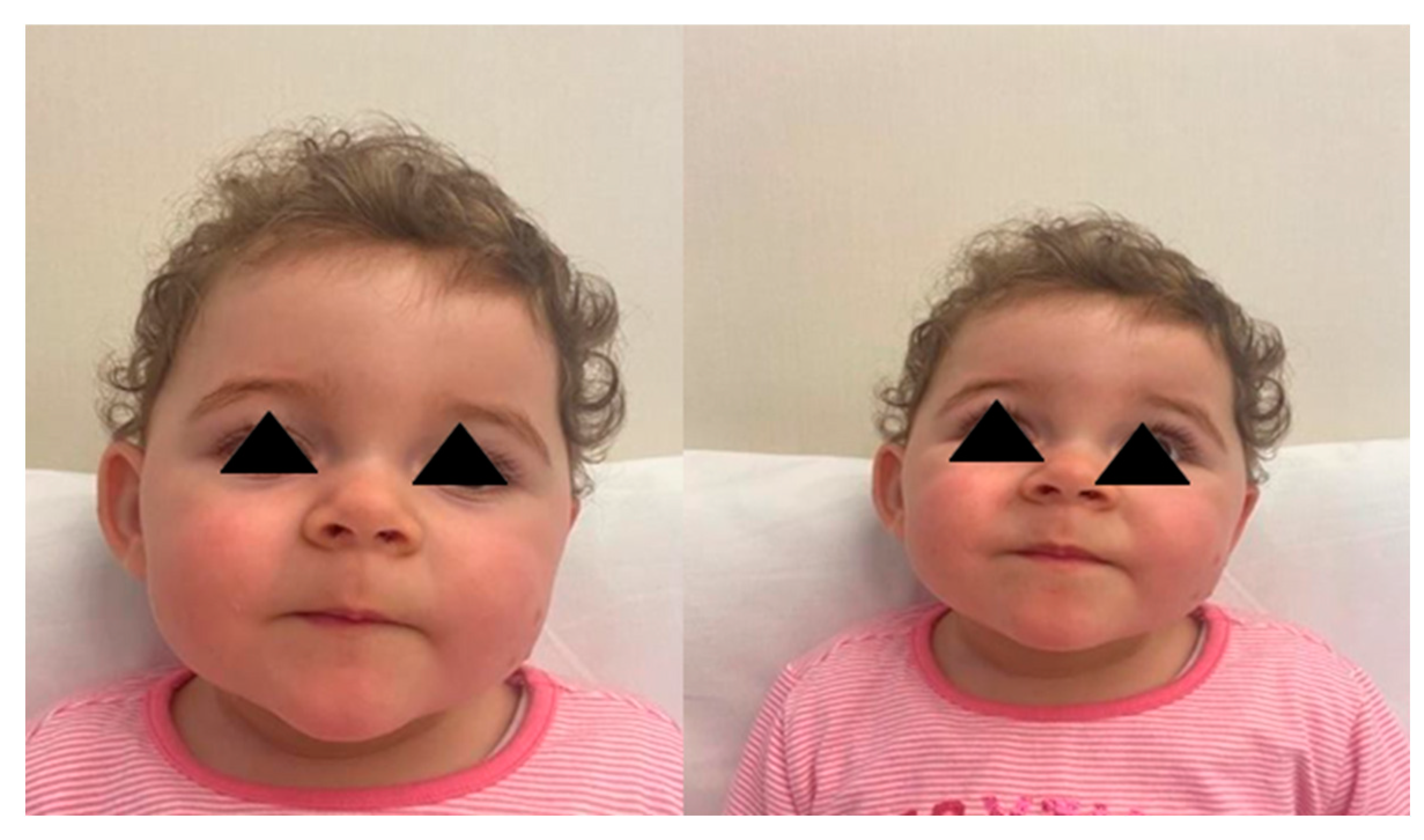
2.1. Case 1
2.2. Case 2
2.3. Psychomotor Development of the Two Babies
2.4. Genetic Analyses of the Two Babies and Their Parents
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, G.R.; Aoto, J.; Tabuchi, K.; Földy, C.; Covy, J.; Yee, A.X.; Wu, D.; Lee, S.-J.; Chen, L.; Malenka, R.C.; et al. Beta-neurexins control neural circuits by regulating synaptic endocannabinoid signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.Y.; Ichtchenko, K.; Sudhof, T.C.; Brose, N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 1100–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, M.S.; Shen, Y.; Tan, W.H.; Jeste, S.S.; Morrow, E.M.; Chen, X.; Mukaddes, N.M.; Yoo, S.Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Dabell, M.P.; Rosenfeld, J.A.; Bader, P.; Escobar, L.F.; El-Khechen, D.; Vallee, S.E.; Dinulos, M.B.P.; Curry, C.; Fisher, J.; Tervo, R.; et al. Investigation of NRXN1 Deletions: Clinical and Molecular Characterization. Am. J. Med. Genet. Part A 2013, 161, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Centanni, T.M.; Green, J.R.; Iuzzini-Seigel, J.; Bartlett, C.W.; Hogan, T.P. Evidence for the multiple hits genetic theory for inherited language impairment: A case study. Front. Genet. 2015, 6, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, B.; Bassett, A.; Chitayat, D.; Chong, K.; Feldman, M.; Flanagan, J.; Goobie, S.; Kawamura, A.; Lowther, C.; Prasad, C.; et al. Deletion of 15q11.2(BP1-BP2) region: Further evidence for lack of phenotypic specificity in a pediatric population. Am. J. Med. Genet. 2015, 167, 2098–2102. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Zahir, F.R.; Baross, A.; Delaney, A.D.; Eydoux, P.; Fernandes, N.D.; Pugh, T.; Marra, M.A.; Friedman, J.M. A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1a. J. Med. Genet. 2007, 45, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Viñas-Jornet, M.; Esteba-Castillo, S.; Gabau, E.; Ribas-Vidal, N.; Baena, N.; San, J.; Ruiz, A.; Coll, M.D.; Novell, R.; Guitart, M. A common cognitive, psychiatric, and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genom. Med. 2014, 2, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez-Wagnera, K.; Jengb, L.J.B.; Slavotineka, A.M.; Sanforda, E.F. 2p16.3 microdeletion with partial deletion of the neurexin-1 gene in a female with developmental delays, short stature, and a congenital diaphragmatic hernia. Clin. Dysmorphol. 2013, 22, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Scibelli, F.; Sinibaldi, L.; Valeri, G.; Caciolo, C.; Novello, R.L.; Novelli, A.; Digilio, M.C.; Tartaglia, M.; Vicari, S. Further insight into the neurobehavioral pattern of children carrying the 2p16.3 heterozygous deletion involving NRXN1: Report of five new cases. Genes Brain Behav. 2020, 19, e12687. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Rees, E.; Walters, J.T.; Escott-Price, V.; Georgieva, L.; Richards, A.L.; Chambert, K.D.; Davies, G.; Legge, S.E.; Moran, J.L.; et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 2014, 75, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béna, F.; Bruno, D.L.; Eriksson, M.; van Ravenswaaij-Arts, C.; Stark, Z.; Dijkhuizen, T.; Gerkes, E.; Gimelli, S.; Ganesamoorthy, D.; Thuresson, A.C.; et al. Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Boone, P.; Sampath, S.; Williams, C.; Bader, P.I.; Mueller, J.M.; Shchelochkov, O.A.; Brown, C.W.; Crawford, H.P.; Phalen, J.; et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet. 2012, 20, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Reissner, C.; Klose, M.; Fairless, R.; Missler, M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc. Natl. Acad. Sci. USA 2008, 105, 15124–15129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Shehhi, M.; Forman, E.B.; Fitzgerald, J.E.; McInerney, V.; Krawczyk, J.; Shen, S.; Betts, D.R.; Mc Ardle, L.; Gorman, K.M.; King, M.D.; et al. NRXN1 deletion syndrome; phenotypic and penetrance data from 34 T families. Eur. J. Med. Genet. 2019, 62, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Castronovo, P.; Baccarin, M.; Ricciardello, A.; Picinelli, C.; Tomaiuolo, P.; Cucinotta, F.; Frittoli, M.; Lintas, C.; Sacco, R.; Persico, A.M. Phenotypic spectrum of NRXN1 mono- and bi-allelic deficiency: A systematic review. Clin. Genet. 2020, 97, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| HINE (Global Score) | Cranial Nerves | Posture | Movements | Muscle Tone | Reflexes and Reactions | |
|---|---|---|---|---|---|---|
| Case 1 | 67 | 14 | 16 | 5 | 18 | 14 |
| Case 2 | 68 | 13 | 18 | 5 | 17 | 15 |
| Case | Source | Genomic Features | Method | Reported Phenotype and Clinical Evaluation |
|---|---|---|---|---|
| Case 1 | F R Zahir et al. [8] | Submicroscopic deletion of chromosome 2p16.3, 320 kb in size, and includes only the part of the NRXN1 gene that codes for the neurexin1a promoter and initial coding exons | Array genomic hybridisation (AGH) | Mild mental retardation, autistic features, multiple vertebral malformations, and an unusual facial appearance |
| Case 2 | Marina Viñas-Jornet et al. [9] | Intragenic 2p16.3 deletion within the NRXN1 | CGH array | Bipolar disorder. IQ of 65. Poor behavioral control, difficulty in acquiring new information, both verbal and visual. Facial dysmorphism: long face, deep-set eyes, hypotelorism, low set ears, prominent premaxilla, a high, narrow palate, and tooth malposition. Dorsal kyphosis and long hands with slender, flexible fingers. |
| Case 3 | Marina Viñas-Jornet et al. [9] | Intragenic 2p16.3 deletion within the NRXN1 | CGH array | Facial dysmorphism, long face, deep-set eyes, hypotelorism, low set ears, prominent premaxilla, and high palate; dorsal kyphosis and finger rigidity. Behavior abnormalities included explosive temper tantrums, violence, and property destruction with a diagnosis of verbal and physically aggressive destructive behavior. IQ of 65 |
| Case 4 | Marina Viñas-Jornet et al. [9] | Intragenic 2p16.3 deletion within the NRXN1 | CGH array | Dysmorphism with a mildly long face, deep-set eyes, prominent premaxilla, and long philtrum. Autistic traits, with hyperactivity and challenging behavior as his most salient psychopathological features. IQ of 53 and a neuropsychological profile characterized by language impairment (both expression and comprehension), poor working memory, and attention. |
| Case 5 | Karla Bermudez- Wagner et al. [10] | 2p16.3 microdeletion with partial deletion of the neurexin-1 gene | CGH array | Morgagni diaphragmatic hernia developmental delays hypotonia, short stature, ptosis, wide mouth, brachydactyly nail hypoplasia |
| Case 6 | Paolo Alfieri et al. [11] | 103.5 Kb deletion at 2p16.3 com- prising one NRXN1 exon | CHG array | Cognitive/developmental delay ASD |
| Case 7 | Paolo Alfieri et al. [11] | Deletion spanning 324.3 Kb including four NRXN1 exons | CGH array | Cognitive/developmental delay ASD |
| Case 8 | Paolo Alfieri et al. [11] | Microdeletion spanning 72 Kb and involving four NRXN1 exons | CGH array | Cognitive/developmental delay |
| Case 9 | Paolo Alfieri et al. [11] | 1.5 Mb deletion, which included three NRXN1 exons | CGH array | Emotional and behavioral problems cognitive/developmental delay ASD |
| Case 10 | Paolo Alfieri et al. [11] | Deletion spanning 144 kb including one NRXN1 intron | CGH array | Cognitive/developmental delay ASD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciacca, M.; Marino, L.; Vitaliti, G.; Falsaperla, R.; Marino, S. NRXN1 Deletion in Two Twins’ Genotype and Phenotype: A Clinical Case and Literature Review. Children 2022, 9, 698. https://doi.org/10.3390/children9050698
Sciacca M, Marino L, Vitaliti G, Falsaperla R, Marino S. NRXN1 Deletion in Two Twins’ Genotype and Phenotype: A Clinical Case and Literature Review. Children. 2022; 9(5):698. https://doi.org/10.3390/children9050698
Chicago/Turabian StyleSciacca, Monica, Lidia Marino, Giovanna Vitaliti, Raffaele Falsaperla, and Silvia Marino. 2022. "NRXN1 Deletion in Two Twins’ Genotype and Phenotype: A Clinical Case and Literature Review" Children 9, no. 5: 698. https://doi.org/10.3390/children9050698
APA StyleSciacca, M., Marino, L., Vitaliti, G., Falsaperla, R., & Marino, S. (2022). NRXN1 Deletion in Two Twins’ Genotype and Phenotype: A Clinical Case and Literature Review. Children, 9(5), 698. https://doi.org/10.3390/children9050698