
Menke–Hennekam Syndrome: A Literature Review and a New Case Report

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
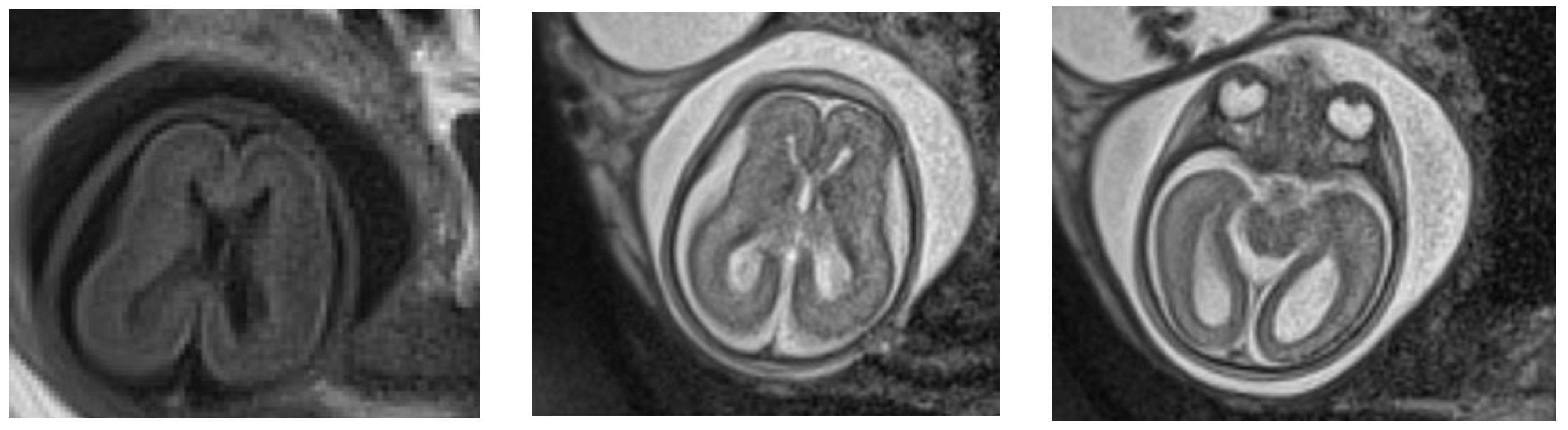
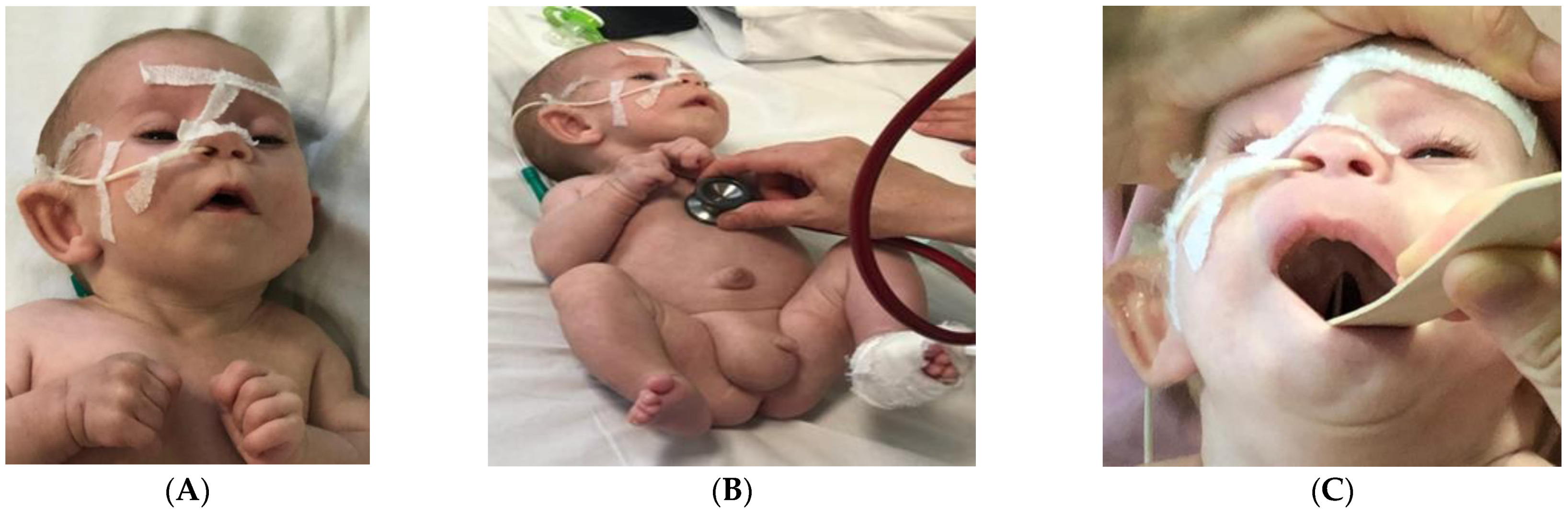
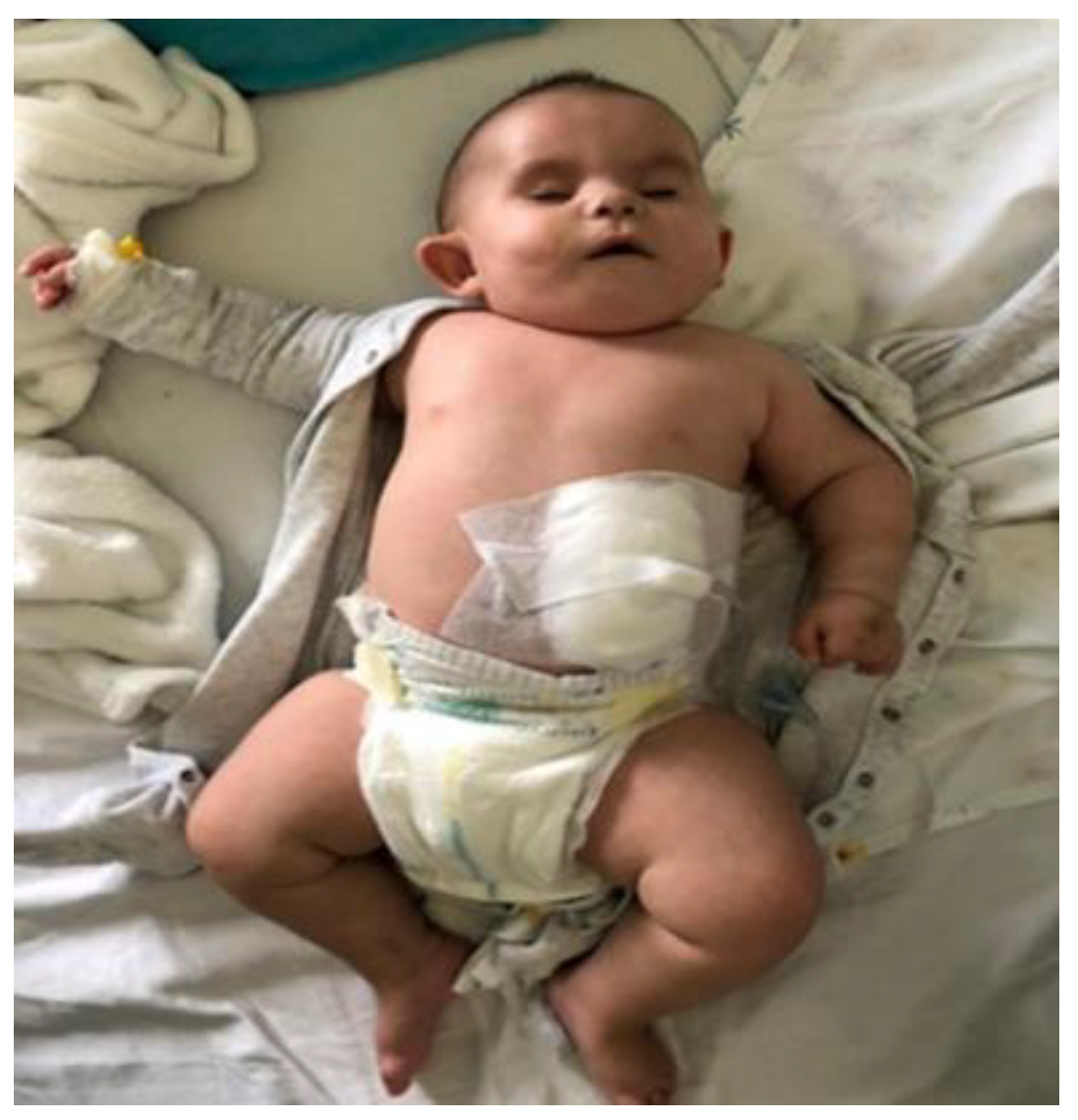
4. Case Report
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dutto, I.; Scalera, C.; Tillhon, M.; Ticli, G.; Passaniti, G.; Cazzalini, O.; Savio, M.; Stivala, L.A.; Gervasini, C.; Larizza, L.; et al. Mutations in CREBBP and EP300 genes affect DNA repair of oxidative damage in Rubinstein-Taybi syndrome cells. Carcinogenesis 2020, 3, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Milani, D.; Manzoni, F.M.; Pezzani, L.; Ajmone, P.; Gervasini, C.; Menni, F.; Esposito, S. Rubinstein-Taybi syndrome: Clinical features, genetic basis, diagnosis, and management. Ital. J. Pediatr. 2015, 41, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menke, L.A.; van Belzen, M.J.; Alders, M.; Cristofoli, F.; DDD Study; Ehmke, N.; Fergelot, P.; Foster, A.; Gerkes, E.H.; Hoffer, M.J.; et al. CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am. J. Med. Genet. A 2016, 170, 2681–2693. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Sayer, R.; Breen, C.; Barton, S.; Pavaine, J.; Sheppard, S.E.; Bedoukian, E.; Skraban, C.; Cuddapah, V.A.; Clayton-Smith, J. Genotype-phenotype specificity in Menke-Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP. Am. J. Med. Genet. A 2019, 179, 1058–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, E.; Takenouchi, T.; Miya, F.; Uehara, T.; Yanagi, K.; Hasegawa, Y.; Ueda, K.; Mizuno, S.; Kaname, T.; Kosaki, K.; et al. The novel and recurrent variants in exon 31 of CREBBP in Japanese patients with Menke-Hennekam syndrome. Am. J. Med. Genet. A 2021, 188, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Kim, Y.; Ryu, H.; Kowall, N.W.; Lee, J.; Ryu, H. Epigenetic mechanisms of Rubinstein-Taybi syndrome. Neuromol. Med. 2014, 16, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef] [PubMed]
- Negri, G.; Gervasini, C. EP300 (E1A binding protein p300). Atlas Genet. Cytogenet. 2017, 19, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Menke, L.A.; DDD Study; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G.; et al. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 2018, 176, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Angius, A.; Uva, P.; Oppo, M.; Persico, I.; Onano, S.; Olla, S.; Pes, V.; Perria, C.; Cuccuru, G.; Atzeni, R.; et al. Confirmation of a new phenotype in an individual with a variant in the last part of exon 30 of CREBBP. Am. J. Med. Genet. A 2019, 179, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Nishi, E.; Ueda, K.; Miya, F.; Yanagi, K.; Mizuno, S.; Kaname, T.; Kosaki, K.; Okamoto, N. The clinical features of individuals of Menke-Hennekam syndrome. Meeting abstract. Eur. J. Hum. Genet. 2020, 28 (Suppl. S1), 461–462. [Google Scholar]
- Menke, L.A.; Jenkins, Z.A.; Williams, E.; Gimenez, G.; O’Neill, A.C.; Hennekam, R.C.; Robertson, S.P. Menke-Hennekam syndrome subtypes caused by variants in the Zinc finger domains ZZ and TAZ2 and the fourth intrinsically disordered linker of CBP and Meeting abstract. Eur. J. Hum. Genet. 2020, 28 (Suppl. S1), 471. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Gene | Variant | Inheritance | Studies |
|---|---|---|---|---|---|
| M1 | M | CREBBP | c.5128T > C (p.Cys1710Arg) | de novo | Menke et al., 2016 [3] |
| M2 | M | CREBBP | c.5240T > G (p.Leu1747Arg) | de novo | |
| M3 | M | CREBBP | c.5357G > C (p.Arg1786Pro) | de novo | |
| M4 | M | CREBBP | c.5456G > T (p.Cys1819Phe) | de novo | |
| M5 | M | CREBBP | c.5478C > G (p.Cys1826Trp) | de novo | |
| M6 | F | CREBBP | c.5513G > A (p.Cys1838Tyr) | de novo | |
| M7 | M | CREBBP | c.5599C > T (p.Arg1867Trp) | de novo | |
| M8 | F | CREBBP | c.5600G > A (p.Arg1867Gln) | de novo | |
| M9 | F | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| M10 | F | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| M11 | F | CREBBP | c.5614A > G (p.Met1872Val) | de novo | |
| M12 | M | CREBBP | c.5155C > G (p.His1719Asp) | de novo | Menke et al., 2018 [11] |
| M13 | M | CREBBP | c.5345C > T (p.Ala1782Val) | de novo | |
| M14 | F | CREBBP | c.5485C > G (p.His1829Asp) | de novo | |
| M15 | F | CREBBP | c.5595_5597del(p.Met1865_Arg1866delinslle) | de novo | |
| M16 | M | CREBBP | c.5600G > A (p.Arg1867Gln) | unknown | |
| M17 | M | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| M18 | M | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| M19 | F | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| M20 | M | CREBBP | c.5603G > A (p.Arg1868Gln) | de novo | |
| M21 | F | CREBBP | c.5608G > C (p.Ala1870Pro) | de novo | |
| M22 | M | CREBBP | c.5614A > G (p.Met1872Val) | de novo | |
| E 1 | F | EP300 | c.5471A > C (p.Gln1824Pro) | de novo | |
| E 2 | F | EP300 | c.5492_5494del (p.Arg181del) | de novo | |
| A1 | M | CREBBP | c.5170G > A (p.Glu1724Lys) | de novo | Angius et al., 2019 [12] |
| B1 | M | CREBBP | c.5357G > A (p.Arg1786His) | de novo | Banka et al., 2019 [4] |
| B2 | F | CREBBP | c.5602C > T (p.Arg1868Trp) | de novo | |
| B3 | F | CREBBP | c.5354G > A (p.Cys1785Try) | de novo | |
| N1 | F | CREBBP | c.5570_5590del | de novo | Nishi et al., 2021 [5] |
| N2 | M | CREBBP | c.5614A > G (p.Met1872Val) | de novo | |
| N3 | M | CREBBP | c.5614A > G (p.Met1872Val) | de novo | |
| N4 | M | CREBBP | c.5991delC (p.Val1998) | de novo | |
| N5 | M | CREBBP | c.6188C > G (p.Ser2063) | de novo | |
| N6 | F | CREBBP | c.6241C > T (p.Gln2081Ter) | de novo |
| Feature | Menke et al. 2018 (n = 24, %) [11] | Banka et al. 2019 (n = 3, %) [4] | Angius et al. 2019 (n = 1, %) [12] | Nishi et al. 2021 (n = 6, %) [5] |
|---|---|---|---|---|
| Intrauterine growth restriction | 7 (29) | 2 (67) | - | 5 (84) |
| Microcephaly | 10/23 (43) | 3 (100) | - | 5 (84) |
| Downslant (D)/upslant (U) palpebral fissures | 3D, 14U (13, 58) | 1D (34) | n.a. | 6U (100) |
| Epicanthus/telecanthus | 13T, 5E (54, 21) | - | 1E (100) | 6E (100) |
| Philtrum long (L)/short (S)/deep (D) | 4S, 12L, 6D (17, 50, 25) | 1L/D (34) | - | 6L (100) |
| Low-set ears | 12 (50) | 2 (67) | 1 (100) | 4 (67) |
| Ptosis (P)/blepharophimosis (B) | 8P, 10B (33, 42) | 1P (34) | - | 1P (17) |
| Hypertelorism | n.a. | 1 (34) | 1 (100) | 6 (100) |
| Depressed nasal bridge | 13 (54) | n.a. | 1 (100) | n.a. |
| Short nose | 12 (50) | 2 (67) | - | 4 (67) |
| Clinodactyly | 6 (25) | - | 1 (100) | 3 (50) |
| Cardiac involvement | 4 (17) | 1 (34) | - | 1 (17) |
| Intellectual impairment | 19/21–24 (80–90) | 3 (100) | 1 (100) | 6 (100) |
| Autistic-like behaviour | 13/20–24 (54–65) | 1 (34) | - | 1 (17) |
| M9 | M10 | B2 | M17 | M18 | M19 | OUR PATIENT | |
|---|---|---|---|---|---|---|---|
| Age at diagnosis (years) | 4 | 0.8 | 0.7 | 2 | 4 | 1 | 0.4 |
| Gender | F | F | F | M | M | F | M |
| Prenatal growth retardation | − | + | + | + | + | + | + |
| Microcephaly | + | + | + | n.a | − | − | + |
| Highly arched eyebrows | + | − | − | n.a | n.a | n.a | + |
| Palpebral fissures upslanted (U)/downslanted (D) | U | U | n.a | U | − | U | U |
| Ptosis(P)/blepharophimosis (B) | P | P | − | P/B | B | P/B | P/B |
| Telecanthus | + | + | n.a | + | + | + | n.a |
| Low set ears | + | + | + | + | + | + | + |
| Short nose | + | + | + | + | + | + | + |
| Long philtrum | + | + | + | + | − | + | + |
| Thin vermilion of upper lip | − | − | n.a | − | + | + | + |
| Cleft palate | n.a | n.a | n.a | − | + | − | + |
| High palate | + | − | n.a | + | + | + | − |
| Severe intellectual disability | + | n.a | + | + | + | n.a | + |
| Cardiac anomalies | − | − | − | − | − | − | atrial septal defect |
| Scoliosis | − | − | + | − | + | +/− | − |
| Age at walking | 7 yr | − | − | − | 4 yr | n.a | − |
| Age at first words | − | − | − | − | − | − | − |
| Syndactyly | + | − | − | + | − | − | + |
| Feeding disorders/gastrostomy (G) | +/G | + | +/G | − | +/G | + | + (G) |
| Hypoacusis | + | + | + | +/− | + | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sima, A.; Smădeanu, R.E.; Simionescu, A.A.; Nedelea, F.; Vlad, A.-M.; Becheanu, C. Menke–Hennekam Syndrome: A Literature Review and a New Case Report. Children 2022, 9, 759. https://doi.org/10.3390/children9050759
Sima A, Smădeanu RE, Simionescu AA, Nedelea F, Vlad A-M, Becheanu C. Menke–Hennekam Syndrome: A Literature Review and a New Case Report. Children. 2022; 9(5):759. https://doi.org/10.3390/children9050759
Chicago/Turabian StyleSima, Aurora, Roxana Elena Smădeanu, Anca Angela Simionescu, Florina Nedelea, Andreea-Maria Vlad, and Cristina Becheanu. 2022. "Menke–Hennekam Syndrome: A Literature Review and a New Case Report" Children 9, no. 5: 759. https://doi.org/10.3390/children9050759
APA StyleSima, A., Smădeanu, R. E., Simionescu, A. A., Nedelea, F., Vlad, A.-M., & Becheanu, C. (2022). Menke–Hennekam Syndrome: A Literature Review and a New Case Report. Children, 9(5), 759. https://doi.org/10.3390/children9050759