
Split Notochord Syndrome with Spinal Column Duplication and Spinal Cord Lipoma: A Case Report


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background and Importance
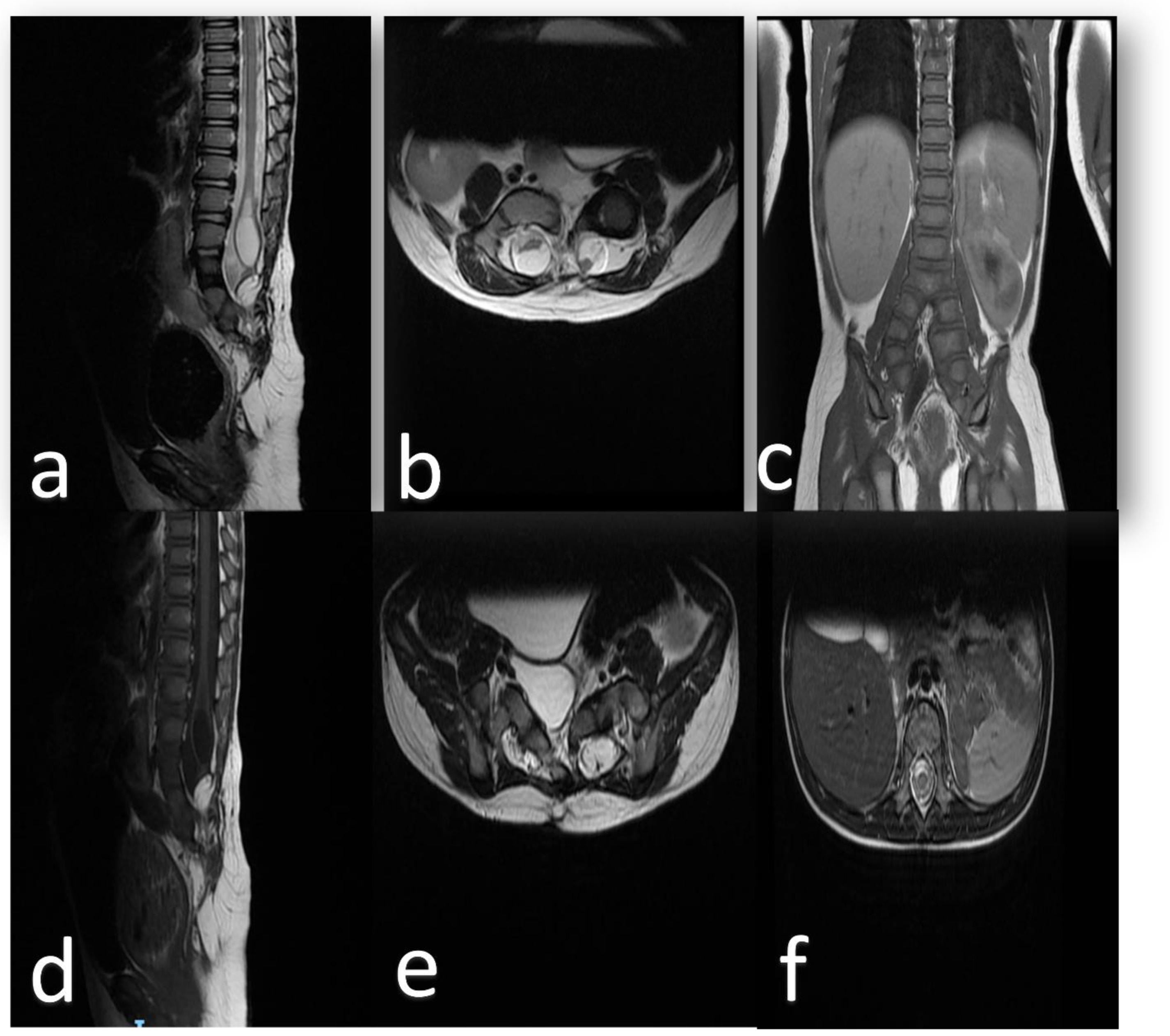
2. Clinical Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bentley, J.F.R.; Smith, J.R. Developmental posterior enteric remnants and spinal malformations: The split notochord syndrome. Arch. Dis. Child. 1960, 35, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, D. Split Cord Malformation: Part II: Clinical Syndrome: Neurosurgery. Neurosurgery 1992, 31, 481–500. [Google Scholar] [CrossRef] [PubMed]
- Wakao, N.; Imagama, S.; Ito, Z.; Ando, K.; Hirano, K.; Tauchi, R.; Muramoto, A.; Matsui, H.; Matsumoto, T.; Matsuyama, Y.; et al. A case of split notochord syndrome: An adult with a spinal endodermal cyst mimicking an intramedullary tumor. Neuropathology 2011, 31, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Raskin, J.S.; Litvack, Z.N.; Selden, N.R. Split Spinal Cord. In Youmans and Winn Neurological Surgery, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 2, pp. 1943–1950. [Google Scholar]
- Jasiewicz, B.; Stachura, M.; Potaczek, T.; Duda, S.; Michno, P.; Kwiatkowski, S. Spine duplication or split notochord syndrome—Case report and literature review. J. Spinal Cord Med. 2018, 43, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.S.; Pang, D. Split Cord Malformations. Neurosurg. Clin. N. Am. 1995, 6, 339–358. [Google Scholar] [CrossRef]
- Pang, D. Split Cord Malformation: Part I: A Unified Theory of Embryogenesis for Double Spinal Cord Malformations. Neurosurgery 1992, 31, 451–480. [Google Scholar] [CrossRef] [PubMed]
- Roelink, H.; Porter, J.A.; Chiang, C.; Tanabe, Y.; Chang, D.T.; Beachy, P.A.; Jessell, T.M. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 1995, 81, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Sarnat, H.; Flores-Sarnat, L. Integrative Classification of Morphology and Molecular Genetics in Central Nervous System Malformations; Academia.edu: San Francisco, CA, USA; Available online: https://www.academia.edu/es/20668358/Integrative_classification_of_morphology_and_molecular_genetics_in_central_nervous_system_malformations (accessed on 14 May 2022).
- Sousa, C.S.M.D.; Castro, B.B.D.; Miranda, C.L.V.M.D.; Bastos, B.B.; Avelino, M.C. Split notochord syndrome: A case in point. Radiol. Bras. 2018, 51, 59–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramzan, M.; Ahmed, S.; Ali, S. Caudal duplication syndrome. J. Clin. Neonatol. 2013, 2, 101. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Bothara, V.P.; Rawat, J.D.; Chaubey, D.; Kumar, P.; Singh, G. A Rare Case of Split Notochord Syndrome. J. Neonatal Surg. 2017, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood-Nash, D.C.; McHugh, K. Diastematomyelia in 172 Children: The Impact of Modern Neuroradiology. Pediatric Neurosurg. 1990, 16, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.C.; Sgouros, S.; Walsh, A.R.; Hockley, A.D. Diastematomyelia in children: Treatment outcome and natural history of associated syringomyelia. Child’s Nerv. Syst. 2006, 23, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Hanif, H.; Khanbabazadeh, S.; Nejat, F.; el Khashab, M. Tethered cord with tandem lipomyelomeningoceles, split cord malformation and thick filum. J. Pediatric Neurosci. 2013, 8, 204. [Google Scholar] [CrossRef] [Green Version]
- Alnefaie, N.; Alharbi, A.; Alamer, O.B.; Khairy, I.; Khairy, S.; Saeed, M.A.; Azzubi, M. Split Cord Malformation: Presentation, Management, and Surgical Outcome. World Neurosurg. 2020, 136, e601–e607. [Google Scholar] [CrossRef] [PubMed]
- Pang, D. Ventral tethering in split cord malformation. Neurosurg. Focus 2001, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.K. Split cord malformation—A study of 300 cases at AIIMS 1990–2006. J. Pediatric Neurosci. 2011, 6 (Suppl. 1), S41. [Google Scholar] [CrossRef] [PubMed]
- Mathkour, M.; Scullen, T.; Huang, B.; Werner, C.; Gouveia, E.E.; Abou-Al-Shaar, H.; Maulucci, C.M.; Steiner, R.B.; St Hilaire, H.; Bui, C.J. Multistage surgical repair for split notochord syndrome with neurenteric fistula: Case report. J. Neurosurg. Pediatrics 2020, 27, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.H.; Dietrich, R.B.; Pais, M.J.; Demos, D.S.; Pribram, H.F. The split notochord syndrome with dorsal enteric fistula. Am. J. Neuroradiol. 1993, 14, 622–627. [Google Scholar] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alelyani, F.; Aronyk, K.; Alghamdi, H.; Alnaami, I. Split Notochord Syndrome with Spinal Column Duplication and Spinal Cord Lipoma: A Case Report. Children 2022, 9, 1138. https://doi.org/10.3390/children9081138
Alelyani F, Aronyk K, Alghamdi H, Alnaami I. Split Notochord Syndrome with Spinal Column Duplication and Spinal Cord Lipoma: A Case Report. Children. 2022; 9(8):1138. https://doi.org/10.3390/children9081138
Chicago/Turabian StyleAlelyani, Fayez, Keith Aronyk, Hashim Alghamdi, and Ibrahim Alnaami. 2022. "Split Notochord Syndrome with Spinal Column Duplication and Spinal Cord Lipoma: A Case Report" Children 9, no. 8: 1138. https://doi.org/10.3390/children9081138