
Development of Monolithic Column Materials for the Separation and Analysis of Glycans


Abstract
:1. Introduction
2. Glycans, Glycosylation and Glycoproteins
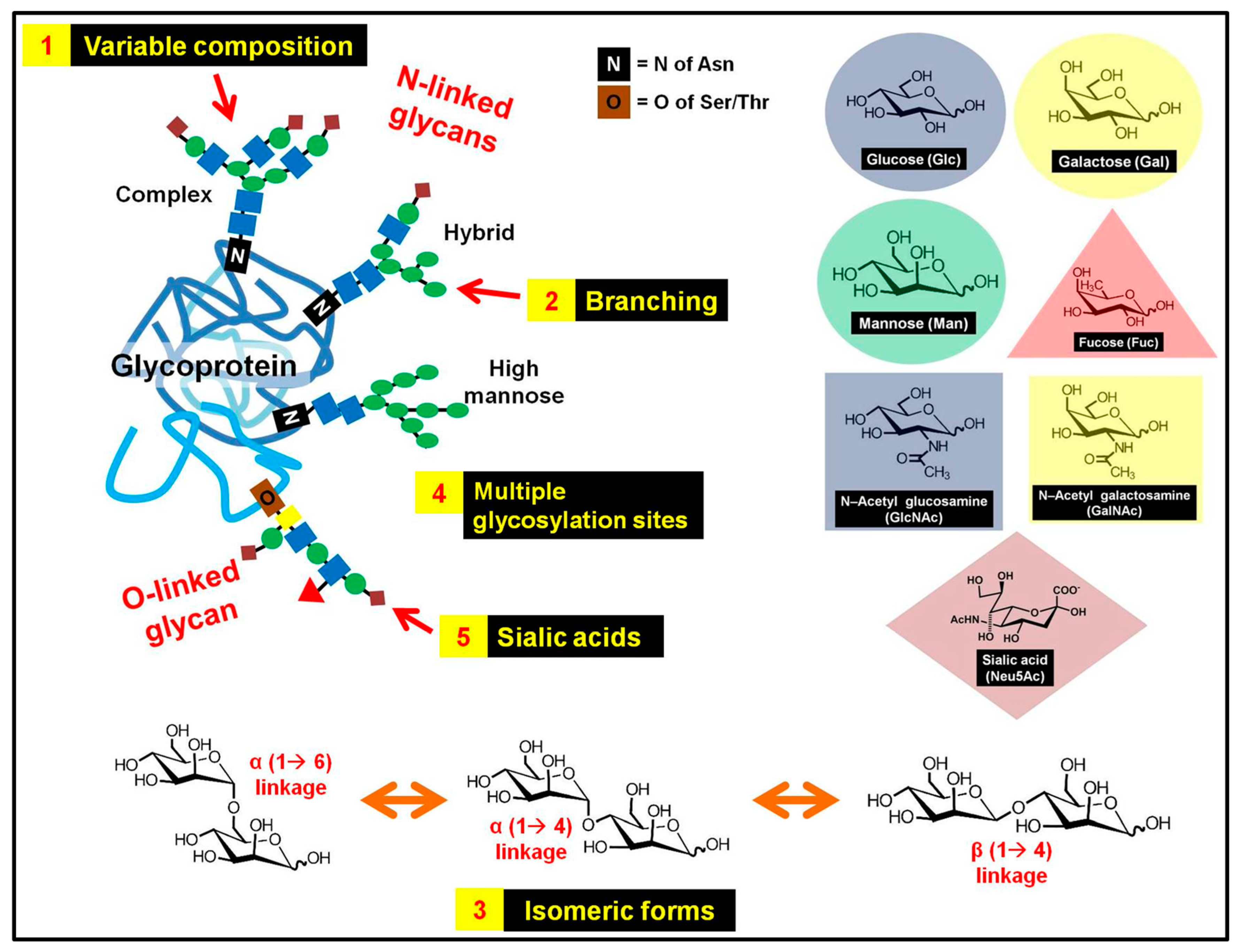
2.1. Complex and Heterogeneous Structure of Glycans

2.2. Approach in Glycomics

3. Monolithic Columns
3.1. Monolithic Columns Can Easily Be Prepared In-Situ

3.2. Monolithic Columns Are Versatile to a Variety of Available Surface Modifications
3.3. Monolithic Columns Have High Permeability and Provide Excellent Mass Transfer with Low Backpressure
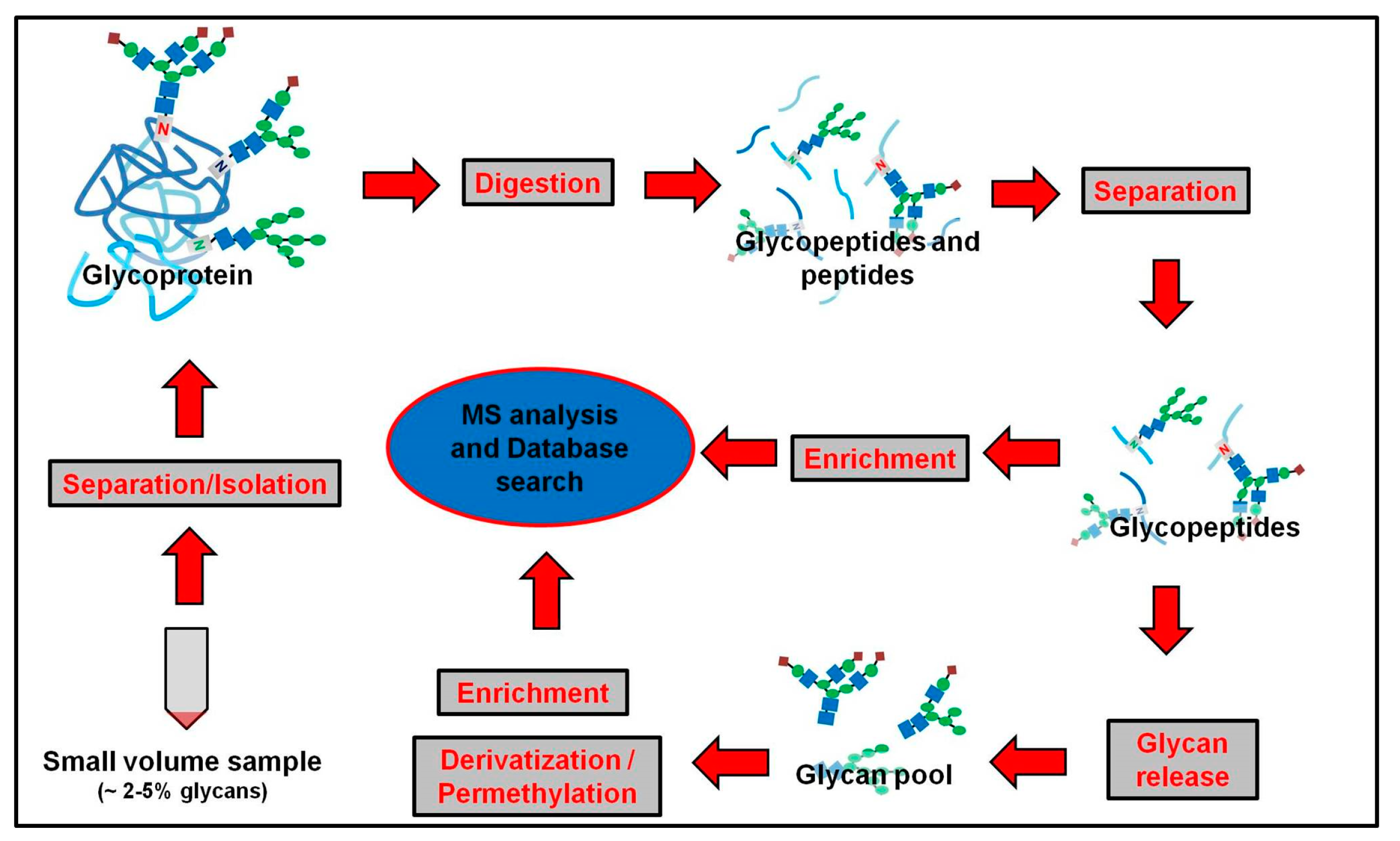
4. Development of Monolithic Column Materials Used in Glycan Release, Separation and Analysis of Glycans
4.1. Monolithic Reactor Columns in Digestion of Glycoproteins and Deglycosylation of Glycopeptides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Column | Immobilization Method | Application | Amount of Enzyme Used * | Reaction Time and Temperature | Stability | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| In-solution | Monolithic reactor | In-solution | Monolithic reactor | |||||
| Trypsin reactor LysC reactor | Via grafted vinyl azlactone (VAL) | Digestion of hIgG Digestion of hIgG | Substrate-to-enzyme ratio of 50:1 (w/w) with 1.25 mg/mL protein | 2.5 mg/mL 0.5 mg/mL | 24 h; 37 °C 24 h; 37 °C | 4 min; 22 °C 6.2 min; 22 °C | 6 months | [82] |
| PNGase F reactor | Via grafted vinyl azlactone (VAL) | Deglycosylation of hIgG integrated on-line with HILIC mode separation and ESI-MS | 0.5 µL | 0.1 µL/min for 2.5 h | 24 h; 37 °C | 5.5 min; room temperature (RT) | 2 months | [83] |
| PNGase F reactor | Via aldehydes (oxidized epoxides) | Simultaneous on-line release and analysis of acidic and neutral N-glycans from 0.1 µL human serum | NS ** | NS ** | Overnight; 37 °C | few min; RT | NS ** | [84] |
| PNGase F micro-reactor | Via direct co-polymerization | Small scale deglycosylation of N-linked glycoproteins | 5 µL of 1 mg/mL | 1 µL of 1 mg/mL | 10 h; 37 °C | 3.5 min; 21–23 °C | 8 weeks | [85] |
| PNGase F reactor | Oriented immobilization via site-specific GSH-GST binding | More efficient deglycosylation of hIgG | 1 mg/mL *** | 1 mg/mL | 2 h; 37 °C *** | 15 s; RT | 5 months | [86] |
4.2. Monolithic Columns in Separation and Enrichment of Glycoproteins, Glycopeptides and Glycans
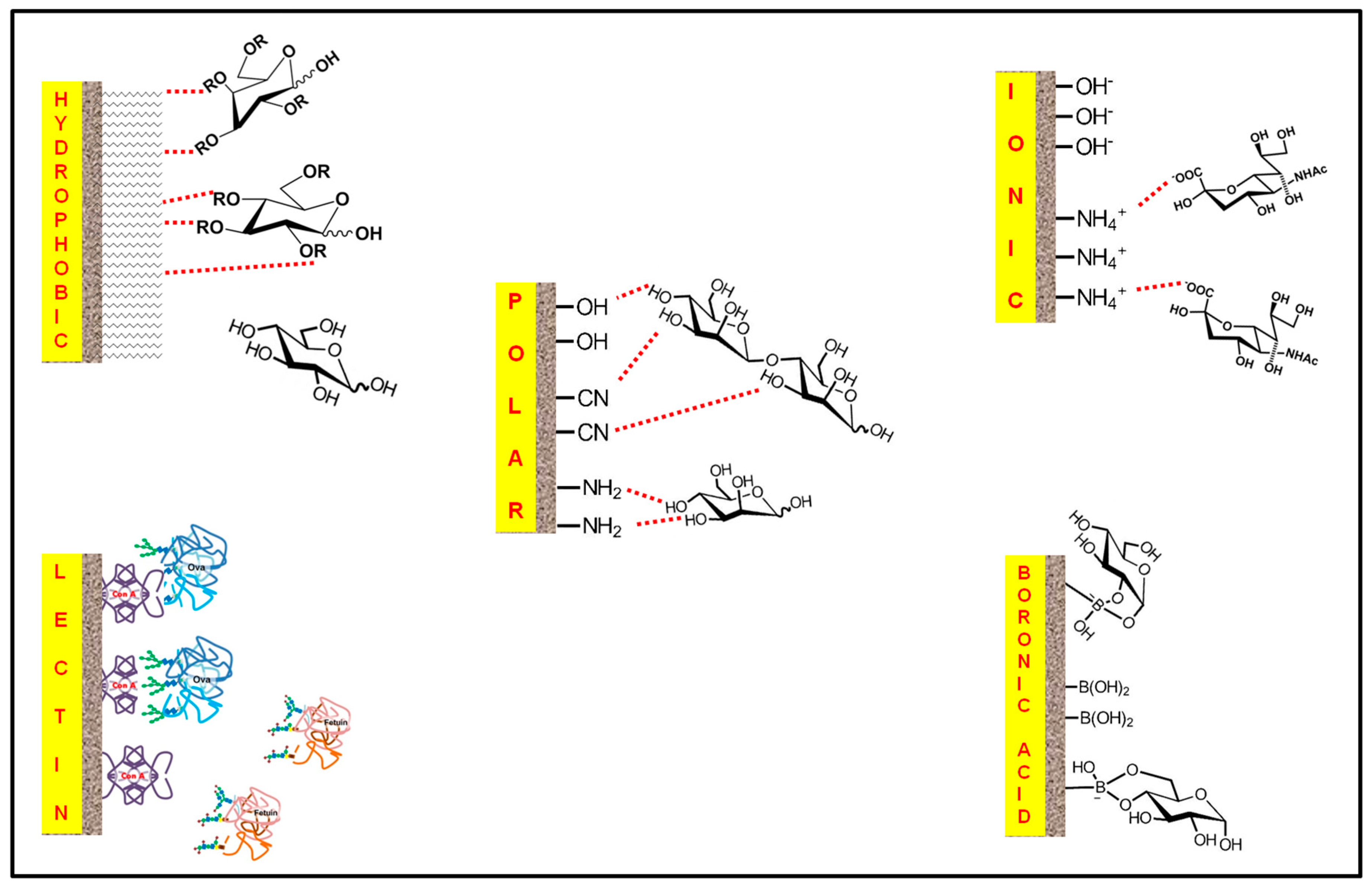
4.2.1. Reverse-Phase Mode

4.2.2. Polar Mode

4.2.3. Electrostatic Mode
4.2.4. Affinity Mode
4.2.4.1. Lectin Affinity
| Lectin | Haptenic sugar | Glycoproteins that binds to lectin | Ref. |
|---|---|---|---|
| Concanavalin A (Con A) | Methyl-α-d-mannopyranoside (Me-α-d-Man) | Ribonuclease B (RNase B) | [98,101] |
| Ovalbumin (Ova) | [101,120] | ||
| Transferrin | [117] | ||
| Horseradish peroxidase (HRP) | [120] | ||
| Wheat germ agglutinin (WGA) | N-Acetyl-d-glucosamine (GlcNAc) | α1-acid glycoprotein (AGP) | [117,119] |
| κ-Casein | [117] | ||
| Fetuin | [119] | ||
| Lens culinaris agglutinin (LCA) | Methyl-α-d-mannopyranoside (Me-α-d-Man) | Glucose oxidase (GOX) | [146] |
| Lipoxidase | [119] | ||
| Human transferrin (HT) | |||
| α1-acid glycoprotein (AGP) | |||
| Avidin | |||
| Collagen | |||
| Erythrina cristagalli lectin (ECL) | Galactose | Desiaylated transferrin | [100] |
| Desiaylated thyroglobulin | |||
| Sambucus Nigra lectin (SNA) | Sialic acid | Fetuin | [124] |
| Oruscomucoid |
| Type of Flow | Functional Ligand | Elution Method (isoc. elu. = isocratic elution; grad. elu. = gradient elution) | Applications | Detection Method | Year & Ref. |
|---|---|---|---|---|---|
| Reverse-phase mode | |||||
| CEC | Alkyl ligands | isoc. elu. 5% ACN | Separation of 2-AB derivatized maltooligosaccharides (Glc4–Glc10) | Laser-induced fluorescence detection | 1997 [87] |
| Polar mode | |||||
| CEC | –OH | isoc. elu. pH 4.5 95% ACN | Separation of nitrophenyl (ortho- or para-) derivatives of mono- and oligosaccharides (βGal and βGlc; βGalNAc and βGlcNAc) | UV detector (304 nm) | 2003 [88] |
| CEC and LC | –OH | isoc. elu. pH 6.0 75% or 65% ACN + 1 mM sulfated β-CD | Profiling of 2AB-derivatized N-glycans derived from Ova and AGP | UV detector (210 nm) | 2009 [89] |
| CEC | –NH2 | isoc. elu. 60% ACN | Separation and analysis of 2-AB derivatized glycans of RNase B | MALDI-TOF MS | 2006 [90] |
| LC | –NH2 | grad. elu. pH 5.5 90%–60% ACN | Separation of underivatized sugars (glucose, maltose, maltotriose, maltotetraose, maltopentaose, maltohexaose, maltoheptaose) Sensitive detection of highly polar components (sucrose, trehalose, maltose and some unknowns) of low volume (50 nL) sample extracts (corn, soybean, Arabidopsis thaliana leaf) | ESI-MS ESI-MS/MS | 2008 [91] |
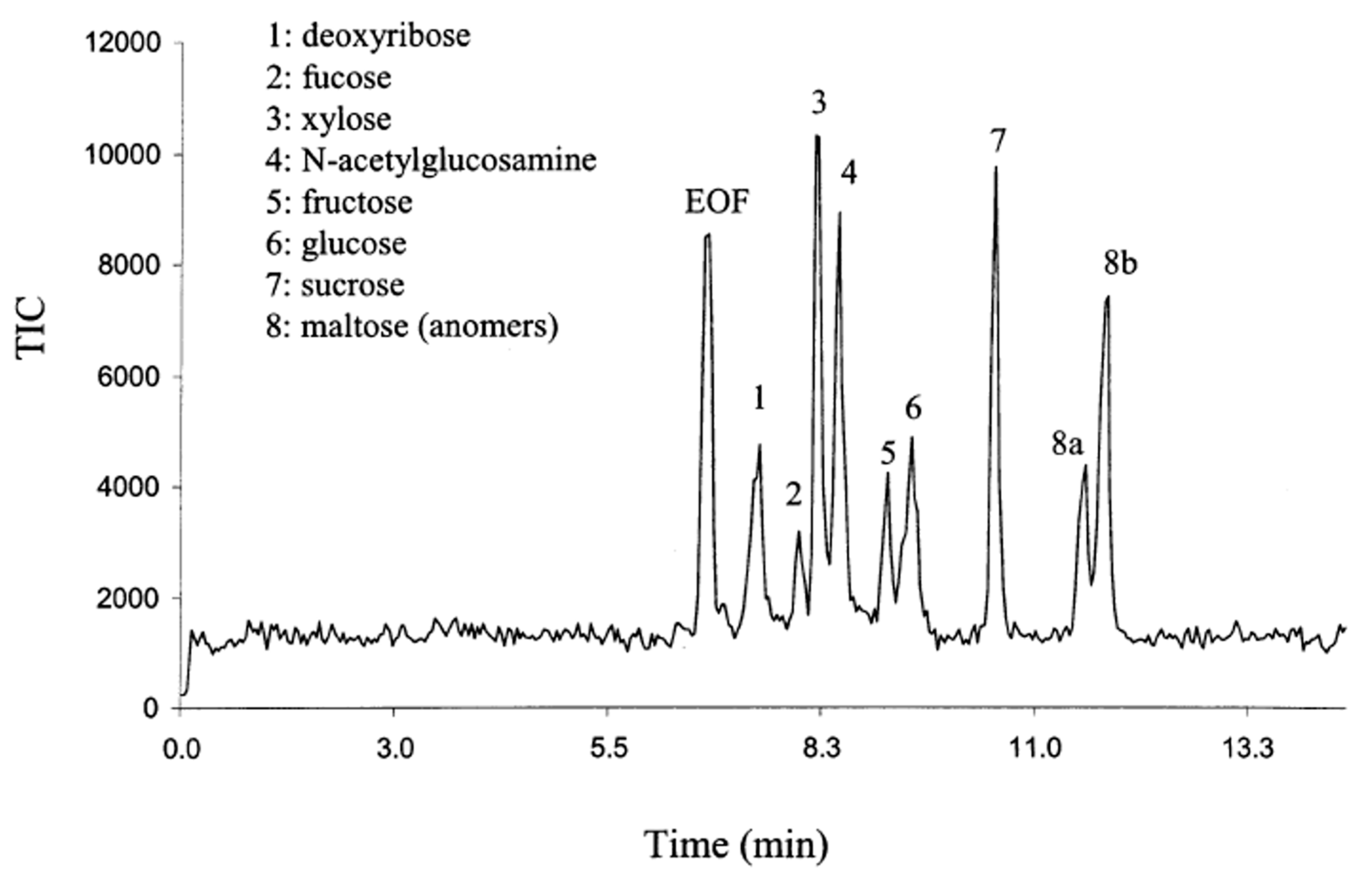
| CEC | –CN | isoc. elu. pH 3.0 (a) 95% or (b) 80% ACN | Separation of femtomole concentration of sugars as alditols:
| ESI-ion trap MS | 2002 [92] |
| CEC | –CN and –NH2 | isoc. elu. pH 3.0 65 or 70 or 75% ACN | Separation of isomeric oligosaccharides:
| ESI-MS/MS | 2002 [93] |
| CEC | –NH2 | isoc. elu. pH 3.0 50% ACN | Separation and analysis of O-glycans from bovine mucin and bile-salt-simulated lipase (BSSL) | FTICR-MS | 2003 [94] |
| Electrostatic mode | |||||
| LC | –OH | grad. elu. pH 7.0 100%–0% KH2PO4 | Separation of IgG from human plasma sample | UV detector (280 nm) | 2009 [95] |
| CEC | –NH2 -OH | grad. elu. increasing amount of NaCl | High resolution profiling of glycoprotein isoforms (AGP, Ova, α-Fetal protein and human glycated hemoglobin) | UV detector | 2011 [96] |
| LC | –NH4+ | isoc. elu. pH 12.8 64 mM NH4OH | Separation of sugars (maltotriose, maltose, and glucose) derived from corn starch | Pulse amperometric detection | 2004 [97] |
| Lectin-affinity mode | |||||
| LC | Con A | isoc. elu. 50% ACN, 2% acetic acid | Enrichment of glycopeptides of trypsin digest of RNase B | ESI-Ion trap -MS/MS | 2006 [98] |
| LC | Con A | Me-α-d-Man | Identification of glycoproteins from 20 µL urine sample | ESI-MS | 2009 [99] |
| LC | ECL | Galactose | Extraction of desialylated transferrin and desialylated thryglobulin from a mixture of proteins (insulin chain B, insulin, cyt c, bovine serum albumin, enolase and carbonic anhydrase) Extraction of spiked desialylated transferrin in E. coli cell lysate | UV Detector | 2011 [100] |
| Manual (96 well) | Con A | Me-α-d-Man | Separation of Ova from BSA | MALDI-TOF | 2012 [101] |
| Boronic acid affinity mode | |||||
| CEC | –B(OH)2 | pH 8.5 ➔ pH 2.7 pH 8.5 ➔ pH 2.7 | Specific capture of Ova from fresh egg white Selective capture of glycoproteins Ovotransferrin (OVT) and Ova from fresh egg white sample | UV detector (214 nm) SDS-PAGE | 2011 [102] |
| CEC | –B(OH)2 | pH 8.5 ➔pH 2.7 | Capture of glycoproteins HRP and lactoferrin from a mixture with non-glycosylated proteins BSA, lactoglobulin, myoglobulin, and cyt c | UV detector (214 nm) | 2009 [103] |
| LC | –B(OH)2 | pH 10.0 ←➔pH 7.4 pH 10.0 ←➔pH 7.4 | Extraction of sialylated glycoprotein human erythropoietin (EPO) from a mixture with non-sialylated glycoprotein HRP and non-glycoprotein BSA Extraction of spiked sialylated glycoprotein EPO from a human serum mixture with non-sialylated glycoprotein RNase B | MALDI-TOF-MS | 2013 [104] |
| CEC | –B(OH)2 | pH 8.0➔pH 3.6 | Selective capture of glycoproteins HRP, Ova from a mixture with non-glycoproteins BSA, bovine hemoglobin (BHb), cyt c, lysozyme and myoglobin Selective capture of glycoproteins OVT and Ova from fresh egg white sample | UV detector (214 nm) SDS-PAGE | 2011 [105] |
| LC | –B(OH)2 | pH 7.2 ➔1% TFA | Identification of glycoproteins Ova, OVT and Ovomucoid (Ovo) | MALDI-TOF-MS | 2013 [106] |
| CEC | –B(OH)2 | pH 7.4➔pH 2.7 pH 7.4➔pH 2.7 | Selective capture of cis-diol containing glycoprotein RNase B and Ova from a mixture with non cis-diol containing glycoprotein RNase A at neutral pH 2D separation of HRP and 2D separation of lactoferrin (showed 2 peaks) | UV detector (214 nm) | 2011 [107] |
| CEC | –B(OH)2 | pH 8.5➔pH 2.7 | Separation of glycoproteins HRP, RNase B and lactoferrin from a mixture with non-glycoproteins myoglobin and BSA | UV detector (214 nm) | 2013 [108] |
| LC | –B(OH)2 | pH 7.0➔pH 2.7 | Specific capture of glycoproteins RNase B, HRP, anti-AFP monoclonal antibody, anti-CEA polyclonal antibody, anti-PSA monoclonal antibody, from a mixture with RNase A, cyt c and β-lactoglobin (possible capture at pH 5.0 was suggested) | UV detector (214 nm) | 2012 [109] |
| Manual (syringe) | –B(OH)2 | pH 9.2 ➔ pH 3.6 | Enrichment of glycopeptides in trypsin digest of HRP Extraction of HRP from a mixture with non-glycosylated BSA via polymer monolith microextraction (PMME) | MALDI-TOF-MS SDS-PAGE | 2009 [110] |
| LC | –B(OH)2 | pH 8.5➔0.2 M HAc | Selective capture of glycoproteins HRP and transferrin from a mixture with non-glycoproteins BSA and cyt c | UV detector (278 nm) | 2011 [111] |
| CEC | –B(OH)2 | pH 8.6➔pH 3.6 | Selective extraction of HRP and enrichment of human serum that contains human serum albumin, IgG, transferrin and spiked HRP | UV detector (214 nm) SDS-PAGE | 2013 [112] |
| LC | –B(OH)2 | pH 7.0➔0.2 M HAc | Selective capture of glycoproteins Ova and OVT from fresh egg white | UV detector (214 nm) SDS-PAGE | 2013 [113] |
| LC/CE | –B(OH)2 | pH 7.0 ➔ pH 2.7 | Rapid selection of HRP-binding DNA aptamers | UV detector (214 nm) CE-LIF | 2013 [114] |
| CEC | –B(OH)2 | pH 7.0 ➔ pH 2.7 | Potential alternative to Protein A in affinity chromatography of glycan-containing antibodies | UV detector MALDI-TOF-MS | 2012 [116] |
| Protein A affinity mode | |||||
| CE | Protein A | pH 7.2 ➔ pH2.3 | Rapid separation of hIgG in human serum | UV detector (239 nm) | 2002 [115] |
| Mixed mode | |||||
| Nano-LC CEC | (1) Lectin affinity (a) WGA (b) Con A (2) Polar (OH–) | (1a) isoc. elu. GlcNAc (1b) isoc. elu. Methyl-α-d-mannopyranoside (2) isoc. elu. 75% ACN with small amount of modifiers β-CD | (1a) Capture of glycoproteins AGP and k-Casein (1b) Capture of glycoproteins Ova and transferrin (2) Polar (CN-OH) based separation of N-glycans derived from AGP and Ova | UV detector (280 nm) | 2009 [117] |
| LC | (1) Hydroph obic Alkyl chain and benzene rings (2) Cationic exchange Negatively charged boronic acid at high pH | grad. elu. Increasing ACN content (20%–40%) in mobile phase with counter-ion (trifluoroacetate anions, TFA) | Separation of iron-binding glycoprotein transferrin from a mixture with non-glycoproteins cyt c and myoglobin | UV detector (214 nm) | 2013 [118] |
4.2.4.2. Boronate Affinity
4.2.5. Mixed-Mode
4.3. Multidimensional Systems
| D | Components | Applications | Detection | Ref. |
|---|---|---|---|---|
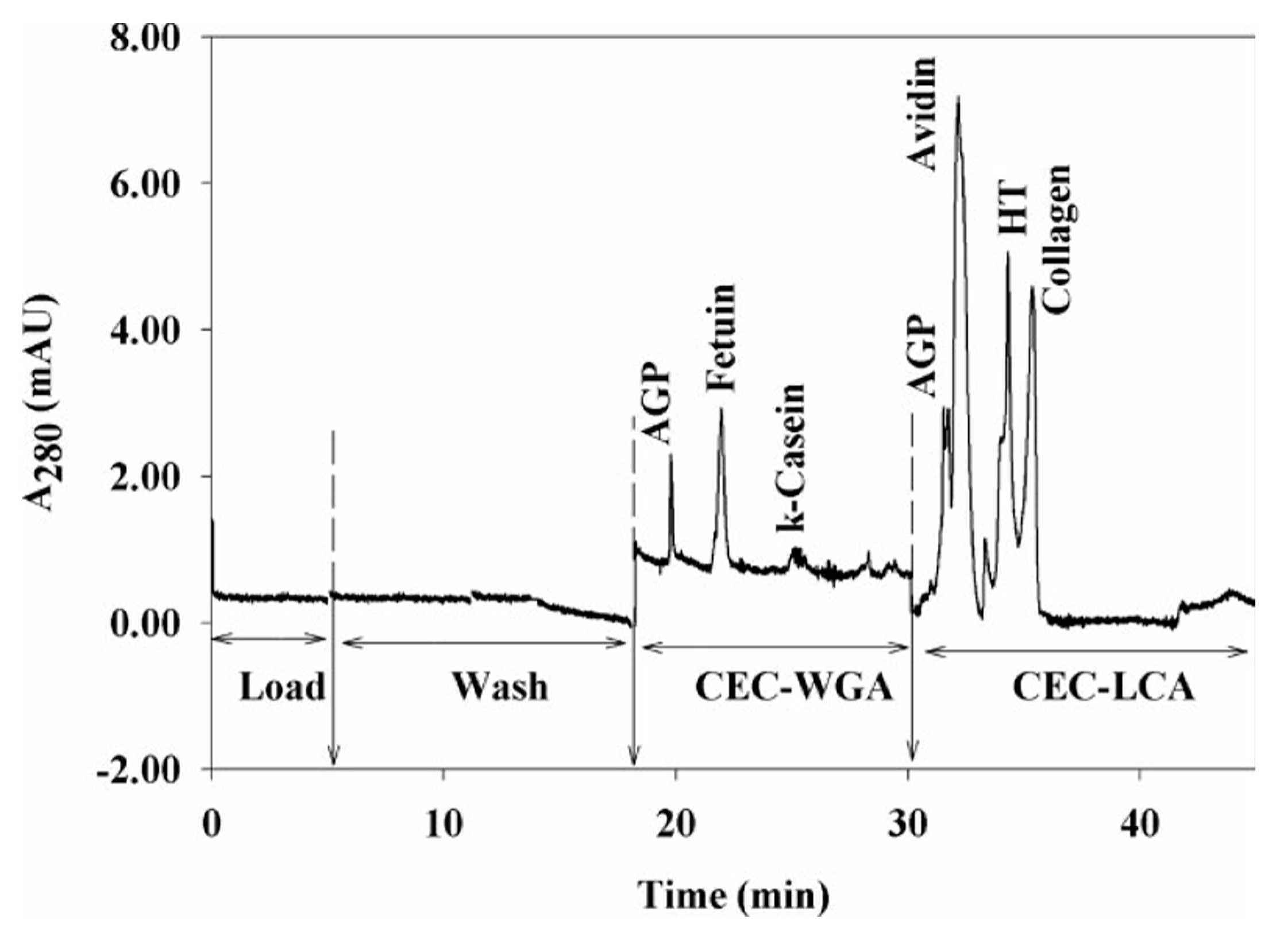
| On-line 2D | (1) LCA affinity column (2) WGA affinity column | Nano-LC and CEC separation of glycoproteins AGP, fetuin, κ-Casein, avidin, holotransferrin, and collagen | UV-detector (280 nm) | 2005 [119] |
| On-line 2D | (1) Con A affinity column (2) RP column | Nano-LC separation of glycoproteins glucose oxidase, human transferrin and OVT from a mixture with non-glycoproteins trypsinogen and α-lactalbumin | UV- detector (210 nm) | 2005 [120] |
| On-line 3D | (1) WGA affinity column (2) Con A affinity column (3) RCI-I affinity column | Capture and profiling breast cancer and disease-free sera glycoproteins | LC-MS/MS | 2012 [121] |
| Off-line 2D | (1) RP column (2) ZIC-HILIC column | Enrichment of glycopeptides using batch HILIC Enhanced separation for profiling and detection of glycopeptides | ESI-MS/MS | 2010 [122] [122] |
| On-line and off-line 2D | (1) PNGase F reactor column (2) HILIC column | Fast and robust analysis of N-glycans from glycoproteins by integrated deglycosylation and enrichment | MALDI-TOF-MS | 2013 [83] |
| On-line 2D | (1) PNGase F reactor column (2) Porous graphitic carbon (PGC) HPLC chip | Online simultaneous release, sample preparation, LC separation and MS analysis of both neutral and acidic N-glycans in just few minutes | LC-MS/MS | 2012 [84] |
| Off-line 2D | (1) RP18e column (2) HILIC column | Glycoproteomic reactor (integrated purification/ desalting, trypsin digestion, enrichment and deglycosylation) | MALDI-TOF-MS | 2013 [123] |


5. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Pfaunmiller, E.L.; Paulemond, M.L.; Dupper, C.M.; Hage, D.S. Affinity monolith chromatography: A review of principles and recent analytical applications. Anal. Bioanal. Chem. 2013, 405, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Walsh, Z.; Paull, B.; Macka, M. Inorganic monoliths in separation science: A review. Anal. Chim. Acta 2012, 750, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Vlakh, E.G.; Tennikova, T.B. Applications of polymethacrylate-based monoliths in high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 2637–2650. [Google Scholar] [CrossRef] [PubMed]
- Potter, O.G.; Hilder, E.F. Porous polymer monoliths for extraction: Diverse applications and platforms. J. Sep. Sci. 2008, 31, 1881–1906. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Huang, X.; Ye, M.; Luo, Q. Monolithic stationary phases for liquid chromatography and capillary electrochromatography. J. Chromatogr. A 2002, 954, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kobayashi, H.; Nakanishi, K.; Minakuchi, H.; Ishizuka, N. Monolithic LC columns. Anal. Chem. 2001, 73, 420a–429a. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.C.; Ghanem, A.; Boon Hon, W.; Hilder, E.F.; Haddad, P.R. Separation and sample pre-treatment in bioanalysis using monolithic phases: A review. Anal. Chim. Acta 2009, 652, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Hofsteenge, J.; Muller, D.R.; de Beer, T.; Loffler, A.; Richter, W.J.; Vliegenthart, J.F. New type of linkage between a carbohydrate and a protein: C-Glycosylation of a specific tryptophan residue in human RNase Us. Biochemistry 1994, 33, 13524–13530. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Peredo, A.; Klein, D.; Macek, B.; Hess, D.; Peter-Katalinic, J.; Hofsteenge, J. C-Mannosylation and o-fucosylation of thrombospondin type 1 repeats. Mol. Cell. Proteomics 2002, 1, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Haynes, P.A. Phosphoglycosylation: A new structural class of glycosylation? Glycobiology 1998, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fasel, N.; Rousseaux, M.; Schaerer, E.; Medof, M.E.; Tykocinski, M.L.; Bron, C. In vitro attachment of glycosyl-inositolphospholipid anchor structures to mouse Thy-1 antigen and human decay-accelerating factor. Proc. Natl. Acad. Sci. USA 1989, 86, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.R.; Davis, J.; Risley, J.M.; Van Etten, R.L. Identification and derivatization of (oligosaccharyl)amines obtained by treatment of asparagine-linked glycopeptides with N-GLYCANASE enzyme. J. Am. Chem. Soc. 1992, 114, 1124–1126. [Google Scholar] [CrossRef]
- Patel, T.; Bruce, J.; Merry, A.; Bigge, C.; Wormald, M.; Parekh, R.; Jaques, A. Use of hydrazine to release in intact and unreduced form both N- and O-linked oligosaccharides from glycoproteins. Biochemistry 1993, 32, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mechref, Y.; Novotny, M.V. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Anal. Chem. 2001, 73, 6063–6069. [Google Scholar] [CrossRef] [PubMed]
- Mechref, Y.; Hu, Y.; Garcia, A.; Hussein, A. Identifying cancer biomarkers by mass spectrometry-based glycomics. Electrophoresis 2012, 33, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Dey, S. Single nucleotide polymorphisms in human P-glycoprotein: Its impact on drug delivery and disposition. Expert Opin. Drug Deliver. 2006, 3, 23–35. [Google Scholar] [CrossRef]
- Greer, D.A.; Ivey, S. Distinct N-glycan glycosylation of P-glycoprotein isolated from the human uterine sarcoma cell line MES-SA/Dx5. Biochim. Biophys. Acta 2007, 1770, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Gribar, J.J.; Ramachandra, M.; Hrycyna, C.A.; Dey, S.; Ambudkar, S.V. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000, 173, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Wanebo, H.J.; Rao, B.; Pinsky, C.M.; Hoffman, R.G.; Stearns, M.; Schwartz, M.K.; Oettgen, H.F. Preoperative carcinoembryonic antigen level as a prognostic indicator in colorectal cancer. N. Engl. J. Med. 1978, 299, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.M.; Pauler, D.K.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Parnes, H.L.; Minasian, L.M.; Ford, L.G.; Lippman, S.M.; Crawford, E.D.; et al. Prevalence of prostate cancer among Men with a prostate-specific antigen level ≤4.0 ng per milliliter. N. Engl. J. Med. 2004, 350, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A. Glycobiology: Toward understanding the function of sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. Three-dimensional mapping of N-linked oligosaccharides using anion-exchange, hydrophobic and hydrophilic interaction modes of high-performance liquid chromatography. J. Chromatogr. A 1996, 720, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, J.; Hashidate, T.; Kasai, K. Glyco-catch method: A lectin affinity technique for glycoproteomics. J. Biomol. Tech. 2002, 13, 205–218. [Google Scholar] [PubMed]
- Woosley, B.; Xie, M.; Wells, L.; Orlando, R.; Garrison, D.; King, D.; Bergmann, C. Comprehensive glycan analysis of recombinant Aspergillus niger endo-polygalacturonase C. Anal. Biochem. 2006, 354, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Ruhaak, L.R.; Zauner, G.; Huhn, C.; Bruggink, C.; Deelder, A.M.; Wuhrer, M. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. [Google Scholar] [CrossRef] [PubMed]
- El Rassi, Z. Recent developments in capillary electrophoresis and capillary electrochromatography of carbohydrate species. Electrophoresis 1999, 20, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shu, C.; Lamb, J.D. High-performance capillary electrophoretic separation of carbohydrates with indirect UV detection using diethylamine and borate as electrolyte additives. J. Capillary Electrophor. 1997, 4, 97–103. [Google Scholar] [PubMed]
- Voegel, P.D.; Zhou, W.; Baldwin, R.P. Integrated capillary electrophoresis/electrochemical detection with metal film electrodes directly deposited onto the capillary tip. Anal. Chem. 1997, 69, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Pilobello, K.T.; Mahal, L.K. Lectin microarrays for glycoprotein analysis. Method. Mol. Biol. 2007, 385, 193–203. [Google Scholar]
- Chen, S.; Haab, B.B. Analysis of glycans on serum proteins using antibody microarrays. Method. Mol. Biol. 2009, 520, 39–58. [Google Scholar]
- Chen, S.; LaRoche, T.; Hamelinck, D.; Bergsma, D.; Brenner, D.; Simeone, D.; Brand, R.E.; Haab, B.B. Multiplexed analysis of glycan variation on native proteins captured by antibody microarrays. Nat. Methods 2007, 4, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Roth, Z.; Yehezkel, G.; Khalaila, I. Identification and quantification of protein glycosylation. Int. J. Carbohydr. Chem. 2012, 2012. Article ID 640923. [Google Scholar]
- Duus, J.Ø.; Gotfredsen, C.H.; Bock, K. Carbohydrate structural determination by NMR spectroscopy: Modern methods and limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef] [PubMed]
- Fellenberg, M.; Behnken, H.N.; Nagel, T.; Wiegandt, A.; Baerenfaenger, M.; Meyer, B. Glycan analysis: Scope and limitations of different techniques—A case for integrated use of LC-MS(/MS) and NMR techniques. Anal. Bioanal. Chem. 2013, 405, 7291–7305. [Google Scholar] [CrossRef] [PubMed]
- Toukach, F.V.; Ananikov, V.P. Recent advances in computational predictions of NMR parameters for the structure elucidation of carbohydrates: Methods and limitations. Chem. Soc. Rev. 2013, 42, 8376–8415. [Google Scholar] [CrossRef] [PubMed]
- Wormald, M.R.; Petrescu, A.J.; Pao, Y.L.; Glithero, A.; Elliott, T.; Dwek, R.A. Conformational studies of oligosaccharides and glycopeptides: Complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 2002, 102, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.; Geyer, R. Strategies for analysis of glycoprotein glycosylation. Biochim. Biophys. Acta 2006, 1764, 1853–1869. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J. Mass Spectrometry and the emerging field of glycomics. Chem. Biol. 2008, 15, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Mechref, Y.; Novotny, M.V. Structural investigations of glycoconjugates at high sensitivity. Chem. Rev. 2002, 102, 321–369. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M. Glycomics using mass spectrometry. Glycoconjug. J. 2013, 30, 11–22. [Google Scholar] [CrossRef]
- Krauss, M.; Singer, H.; Hollender, J. LC-high resolution MS in environmental analysis: From target screening to the identification of unknowns. Anal. Bioanal. Chem. 2010, 397, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; Lyndon, M.M.; Dixon, R.B.; Muddiman, D.C. Mass measurement accuracy comparisons between a double-focusing magnetic sector and a time-of-flight mass analyzer. Rapid Commun. Mass Spectrom. 2008, 22, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.H.; Cooks, R.G.; Noll, R.J. Orbitrap mass spectrometry: Instrumentation, ion motion and applications. Mass Spectrom. Rev. 2008, 27, 661–699. [Google Scholar] [CrossRef] [PubMed]
- March, R.E. An Introduction to quadrupole ion trap mass spectrometry. J. Mass Spectrom. 1997, 32, 351–369. [Google Scholar] [CrossRef]
- Krutchinsky, A.N.; Kalkum, M.; Chait, B.T. Automatic identification of proteins with a MALDI-quadrupole ion trap mass spectrometer. Anal. Chem. 2001, 73, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Trauger, S.A.; Webb, W.; Siuzdak, G. Peptide and protein analysis with mass spectrometry. Spectroscopy 2002, 16, 15–28. [Google Scholar] [CrossRef]
- Boesl, U.; Weinkauf, R.; Schlag, E.W. Reflectron time-of-flight mass spectrometry and laser excitation for the analysis of neutrals, ionized molecules and secondary fragments. Int. J. Mass Spectrom. 1992, 112, 121–166. [Google Scholar] [CrossRef]
- Atwood, J.A.; Cheng, L.; Alvarez-Manilla, G.; Warren, N.L.; York, W.S.; Orlando, R. Quantitation by isobaric labeling: Applications to glycomics. J. Proteome Res. 2007, 7, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Hopfgartner, G.; Varesio, E.; Tschappat, V.; Grivet, C.; Bourgogne, E.; Leuthold, L.A. Triple quadrupole linear ion trap mass spectrometer for the analysis of small molecules and macromolecules. J. Mass Spectrom. 2004, 39, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Cravatt, B.F.; Siuzdak, G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem 2005, 6, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.J.; Xie, H.; Bandhakavi, S.; Popko, J.; Mohan, A.; Carlis, J.V.; Higgins, L. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J. Proteome Res. 2007, 6, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- Pabst, M.; Bondili, J.S.; Stadlmann, J.; Mach, L.; Altmann, F. Mass + retention time = structure: A strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans. Anal. Chem. 2007, 79, 5051–5057. [Google Scholar] [CrossRef] [PubMed]
- Harvey, D.J. Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: An update for 2009–2010. Mass Spectrom. Rev. 2014. [Google Scholar] [CrossRef]
- Harvey, D.J. Identification of protein-bound carbohydrates by mass spectrometry. Proteomics 2001, 1, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Deelder, A.M. Negative-mode MALDI-TOF/TOF-MS of oligosaccharides labeled with 2-aminobenzamide. Anal. Chem. 2005, 77, 6954–6959. [Google Scholar] [CrossRef] [PubMed]
- Mechref, Y.; Kang, P.; Novotny, M.V. Differentiating structural isomers of sialylated glycans by matrix-assisted laser desorption/ionization time-of-flight/time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Catalina, M.I.; Deelder, A.M.; Hokke, C.H. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J. Chromatogr. B 2007, 849, 115–128. [Google Scholar] [CrossRef]
- Prien, J.M.; Ashline, D.J.; Lapadula, A.J.; Zhang, H.; Reinhold, V.N. The high mannose glycans from bovine ribonuclease B isomer characterization by ion trap MS. J. Am. Soc. Mass Spectrom. 2009, 20, 539–556. [Google Scholar] [CrossRef] [PubMed]
- An, H.J.; Lebrilla, C.B. Structure elucidation of native N- and O-linked glycans by tandem mass spectrometry (tutorial). Mass Spectrom. Rev. 2011, 30, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; An, H.J.; Hedrick, J.L.; Lebrilla, C.B. Collision-induced dissociation tandem mass spectrometry for structural elucidation of glycans. Method. Mol. Biol. 2009, 534, 133–145. [Google Scholar]
- Jonscher, K.R.; Yates, J.R., 3rd. The quadrupole ion trap mass spectrometer—A small solution to a big challenge. Anal. Biochem. 1997, 244, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scigelova, M.; Hornshaw, M.; Giannakopulos, A.; Makarov, A. Fourier transform mass spectrometry. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Bahr, U.; Pfenninger, A.; Karas, M.; Stahl, B. High-sensitivity analysis of neutral underivatized oligosaccharides by nanoelectrospray mass spectrometry. Anal. Chem. 1997, 69, 4530–4535. [Google Scholar] [CrossRef] [PubMed]
- Alley, W.R., Jr.; Mechref, Y.; Novotny, M.V. Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun. Mass Spectrom. 2009, 23, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J. Mass spectrometry and the emerging field of glycomics. Chem. Biol. 2008, 15, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, N.; Zaia, J. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal. Chem. 2012, 84, 3040–3048. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.M.; Lazar, A.C.; Cortes, D.F.; Kabulski, J.L. Recent advances in the MS analysis of glycoproteins: Theoretical considerations. Electrophoresis 2011, 32, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M. Glycomics using mass spectrometry. Glycoconjug. J. 2013, 30, 11–22. [Google Scholar] [CrossRef]
- Cortes, D.F.; Kabulski, J.L.; Lazar, A.C.; Lazar, I.M. Recent advances in the MS analysis of glycoproteins: Capillary and microfluidic workflows. Electrophoresis 2011, 32, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Pabst, M.; Altmann, F. Glycan analysis by modern instrumental methods. Proteomics 2011, 11, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Luke, B.; Andresson, T.; Blonder, J. 18O stable isotope labeling in MS-based proteomics. Brief. Funct. Genomic. Proteomic. 2009, 8, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Saito, H.; Yamauchi, Y.; Shinkawa, T.; Taoka, M.; Hirabayashi, J.; Kasai, K.; Takahashi, N.; Isobe, T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol. 2003, 21, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, G.; Melo-Braga, M.N.; Engholm-Keller, K.; Parker, B.L.; Larsen, M.R. Chemical deamidation: A common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J. Proteome Res. 2012, 11, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Ren, Y.; Alpert, A.J.; Sze, S.K. Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.M.; Lim, J.M.; Wells, L.; Bergmann, C.; Orlando, R. A potential pitfall in 18O-based N-linked glycosylation site mapping. Rapid Commun. Mass Spectrom. 2007, 21, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Deelder, A.M.; Hokke, C.H. Protein glycosylation analysis by liquid chromatography-mass spectrometry. J. Chromatogr. B 2005, 825, 124–133. [Google Scholar] [CrossRef]
- Mechref, Y. Analysis of glycans derived from glycoconjugates by capillary electrophoresis-mass spectrometry. Electrophoresis 2011, 32, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Tennikova, T.B.; Bleha, M.; Švec, F.; Almazova, T.V.; Belenkii, B.G. High-performance membrane chromatography of proteins, a novel method of protein separation. J. Chromatogr. A 1991, 555, 97–107. [Google Scholar] [CrossRef]
- Svec, F.; Frechet, J.M.J. Continuous rods of macroporous polymer as high-performance liquid chromatography separation media. Anal. Chem. 1992, 64, 820–822. [Google Scholar] [CrossRef]
- Hjertén, S.; Liao, J.-L.; Zhang, R. High-performance liquid chromatography on continuous polymer beds. J. Chromatogr. A 1989, 473, 273–275. [Google Scholar] [CrossRef]
- Krenkova, J.; Lacher, N.A.; Svec, F. Highly efficient enzyme reactors containing trypsin and endoproteinase LysC immobilized on porous polymer monolith coupled to MS suitable for analysis of antibodies. Anal. Chem. 2009, 81, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Krenkova, J.; Lacher, N.A.; Svec, F. Multidimensional system enabling deglycosylation of proteins using a capillary reactor with peptide-N-glycosidase F immobilized on a porous polymer monolith and hydrophilic interaction liquid chromatography-mass spectrometry of glycans. J. Chromatogr. A 2009, 1216, 3252–3259. [Google Scholar] [CrossRef] [PubMed]
- Jmeian, Y.; Hammad, L.A.; Mechref, Y. Fast and efficient online release of N-glycans from glycoproteins facilitating liquid chromatography-tandem mass spectrometry glycomic profiling. Anal. Chem. 2012, 84, 8790–8796. [Google Scholar] [CrossRef] [PubMed]
- Palm, A.K.; Novotny, M.V. A monolithic PNGase F enzyme microreactor enabling glycan mass mapping of glycoproteins by mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Krenkova, J.; Szekrenyes, A.; Keresztessy, Z.; Foret, F.; Guttman, A. Oriented immobilization of peptide-N-glycosidase F on a monolithic support for glycosylation analysis. J. Chromatogr. A 2013, 1322, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Palm, A.; Novotny, M.V. Macroporous Polyacrylamide/Poly(ethylene glycol) Matrixes as Stationary Phases in Capillary Electrochromatography. Anal. Chem. 1997, 69, 4499–4507. [Google Scholar] [CrossRef]
- Allen, D.; El Rassi, Z. Capillary electrochromatography with monolithic silica columns III. Preparation of hydrophilic silica monoliths having surface-bound cyano groups: Chromatographic characterization and application to the separation of carbohydrates, nucleosides, nucleic acid bases and other neutral polar species. J. Chromatogr. A 2004, 1029, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; El Rassi, Z. Neutral polar methacrylate-based monoliths for normal phase nano-LC and CEC of polar species including N-glycans. J. Sep. Sci. 2009, 32, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Guryča, V.; Mechref, Y.; Palm, A.K.; Michálek, J.; Pacáková, V.; Novotný, M.V. Porous polyacrylamide monoliths in hydrophilic interaction capillary electrochromatography of oligosaccharides. J. Biochem. Biophys. Method. 2007, 70, 3–13. [Google Scholar] [CrossRef]
- Ikegami, T.; Horie, K.; Saad, N.; Hosoya, K.; Fiehn, O.; Tanaka, N. Highly efficient analysis of underivatized carbohydrates using monolithic-silica-based capillary hydrophilic interaction (HILIC) HPLC. Anal. Bioanal. Chem. 2008, 391, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Que, A.H.; Novotny, M.V. Separation of neutral saccharide mixtures with capillary electrochromatography using hydrophilic monolithic columns. Anal. Chem. 2002, 74, 5184–5191. [Google Scholar] [CrossRef] [PubMed]
- Que, A.H.; Novotny, M.V. Structural characterization of neutral oligosaccharide mixtures through a combination of capillary electrochromatography and ion trap tandem mass spectrometry. Anal. Bioanal. Chem. 2003, 375, 599–608. [Google Scholar] [PubMed]
- Que, A.H.; Mechref, Y.; Huang, Y.; Taraszka, J.A.; Clemmer, D.E.; Novotny, M.V. Coupling capillary electrochromatography with electrospray Fourier transform mass spectrometry for characterizing complex oligosaccharide pools. Anal. Chem. 2003, 75, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Yang, G.-L.; Bai, L.-G.; Feng, X.-J.; Yang, G.-Q.U.N. Separation of immunoglobulin G and immunoglobulin Y on poly(vinyl ester resin-co-ethylene dimethacrylate) monolith. Chin. J. Anal. Chem. 2009, 37, 325–329. [Google Scholar] [CrossRef]
- Liu, J.; Ren, L.; Liu, Y.; Li, H.; Liu, Z. Weak anion exchange chromatographic profiling of glycoprotein isoforms on a polymer monolithic capillary. J. Chromatogr. A 2012, 1228, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Hilder, E.F.; Svec, F.; Fréchet, J.M.J. Latex-functionalized monolithic columns for the separation of carbohydrates by micro anion-exchange chromatography. J. Chromatogr. A 2004, 1053, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Bedair, M.; Oleschuk, R.D. Lectin affinity chromatography using porous polymer monolith assisted nanoelectrospray MS/MS. Analyst 2006, 131, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, N.; Pennathur, S.; Goodison, S.; Lubman, D.M. Enrichment of glycoproteins using nanoscale chelating concanavalin a monolithic capillary chromatography. Anal. Chem. 2009, 81, 3776–3783. [Google Scholar] [CrossRef] [PubMed]
- Alwael, H.; Connolly, D.; Clarke, P.; Thompson, R.; Twamley, B.; O'Connor, B.; Paull, B. Pipette-tip selective extraction of glycoproteins with lectin modified gold nano-particles on a polymer monolithic phase. Analyst 2011, 136, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichelt, S.; Elsner, C.; Prager, A.; Naumov, S.; Kuballa, J.; Buchmeiser, M.R. Amino-functionalized monolithic spin-type columns for high-throughput lectin affinity chromatography of glycoproteins. Analyst 2012, 137, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.A.; Pang, J.L.; Lin, Y.; Huang, H.; Cai, Z.W.; Zhang, L.; Chen, G.N. Preparation and evaluation of a phenylboronate affinity monolith for selective capture of glycoproteins by capillary liquid chromatography. Analyst 2011, 136, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Liu, Y.; Dong, M.; Liu, Z. Synthesis of hydrophilic boronate affinity monolithic capillary for specific capture of glycoproteins by capillary liquid chromatography. J. Chromatogr. A 2009, 1216, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Bie, Z.; Liu, Y.; Liu, Z. Fine-tuning the specificity of boronate affinity monoliths toward glycoproteins through pH manipulation. Analyst 2013, 138, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Pang, J.; Yang, H.; Cai, Z.; Zhang, L.; Chen, G. One-pot synthesis of an organic-inorganic hybrid affinity monolithic column for specific capture of glycoproteins. Chem. Commun. (Camb) 2011, 47, 9675–9677. [Google Scholar] [CrossRef]
- Wang, S.T.; Chen, D.; Ding, J.; Yuan, B.F.; Feng, Y.Q. Borated titania, a new option for the selective enrichment of cis-diol biomolecules. Chemistry 2013, 19, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ren, L.; Liu, Z. A unique boronic acid functionalized monolithic capillary for specific capture, separation and immobilization of cis-diol biomolecules. Chem. Commun. (Camb) 2011, 47, 5067–5069. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Ren, L.; Li, H.; Liu, Z. Development of poly((3-acrylamidophenyl)boronic acid-co-N,N-methylenebisacrylamide) monolithic capillary for the selective capture of cis-diol biomolecules. Anal. Method. 2013, 5, 5444–5449. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Liu, Y.; Liu, Z. A benzoboroxole-functionalized monolithic column for the selective enrichment and separation of cis-diol containing biomolecules. Chem. Commun. (Camb) 2012, 48, 4115–4117. [Google Scholar] [CrossRef]
- Chen, M.; Lu, Y.; Ma, Q.; Guo, L.; Feng, Y.Q. Boronate affinity monolith for highly selective enrichment of glycopeptides and glycoproteins. Analyst 2009, 134, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lin, Z.; He, X.; Chen, L.; Zhang, Y. Synthesis and application of a macroporous boronate affinity monolithic column using a metal-organic gel as a porogenic template for the specific capture of glycoproteins. J. Chromatogr. A 2011, 1218, 9194–9201. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wang, J.; Tan, X.; Sun, L.; Yu, R.; Yang, H.; Chen, G. Preparation of boronate-functionalized molecularly imprinted monolithic column with polydopamine coating for glycoprotein recognition and enrichment. J. Chromatogr. A 2013, 1319, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Mao, J.; He, X.W.; Chen, L.X.; Zhang, Y.K. Preparation of a boronate-functionalized affinity hybrid monolith for specific capture of glycoproteins. Anal. Bioanal. Chem. 2013, 405, 5321–5331. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Chen, Y.; Lu, C.; Liu, Z. Efficient selection of glycoprotein-binding DNA aptamers via boronate affinity monolithic capillary. Anal. Chem. 2013, 85, 8277–8283. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zou, H.; Mo, W.; Huang, X.; Wu, R. Protein A immobilized monolithic capillary column for affinity chromatography. Anal. Chim. Acta 2002, 466, 141–150. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Liu, Z. Restricted access boronate affinity porous monolith as a protein A mimetic for the specific capture of immunoglobulin G. Chem. Sci. 2012, 3, 1467–1471. [Google Scholar] [CrossRef]
- Zhong, H.; El Rassi, Z. Monolithic silica capillary columns having immobilized lectins and surface bound polar functionalities for lectin affinity and normal phase nano-LC and CEC of glycoconjugates, respectively. J. Sep. Sci. 2009, 32, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Aydoğan, C.; Yılmaz, F.; Denizli, A. Cation exchange/hydrophobic interaction monolithic chromatography of small molecules and proteins by nano liquid chromatography. J. Sep. Sci. 2013, 36, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Okanda, F.M.; El Rassi, Z. Affinity monolithic capillary columns for glycomics/proteomics: 1. Polymethacrylate monoliths with immobilized lectins for glycoprotein separation by affinity capillary electrochromatography and affinity nano-liquid chromatography in either a single column or columns coupled in series. Electrophoresis 2006, 27, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Bedair, M.; El Rassi, Z. Affinity chromatography with monolithic capillary columns. II. Polymethacrylate monoliths with immobilized lectins for the separation of glycoconjugates by nano-liquid affinity chromatography. J. Chromatogr. A 2005, 1079, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; El Rassi, Z. Tandem lectin affinity chromatography monolithic columns with surface immobilised concanavalin A, wheat germ agglutinin and Ricinus communis agglutinin-I for capturing sub-glycoproteomics from breast cancer and disease-free human sera. J. Sep. Sci. 2012, 35, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, J.; Karas, M.; Jiang, W.; Hendriks, R.; Andrecht, S. Enhanced glyco-profiling by specific glycopeptide enrichment and complementary monolithic nano-LC (ZIC-HILIC/RP18e)/ESI-MS analysis. J. Sep. Sci. 2010, 33, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F.; Lin, H.; Zhu, J.; Bian, Y.; Cheng, K.; Zou, H. Monolithic capillary column based glycoproteomic reactor for high-sensitive analysis of N-glycoproteome. Anal. Chem. 2013, 85, 2847–2852. [Google Scholar] [CrossRef] [PubMed]
- Madera, M.; Mechref, Y.; Klouckova, I.; Novotny, M.V. Semiautomated high-sensitivity profiling of human blood serum glycoproteins through lectin preconcentration and multidimensional chromatography/tandem mass spectrometry. J. Proteome Res. 2006, 5, 2348–2363. [Google Scholar] [CrossRef] [PubMed]
- Hjerten, S. "Molecular sieve" chromatography on polyacrylamide gels, prepared according to a simplified method. Arch. Biochem. Biophys. 1962, Suppl 1, 147–151. [Google Scholar]
- Minakuchi, H.; Nakanishi, K.; Soga, N.; Ishizuka, N.; Tanaka, N. Octadecylsilylated porous silica rods as separation media for reversed-phase liquid chromatography. Anal. Chem. 1996, 68, 3498–3501. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sakai-Kato, K.; Toyo'oka, T. Silica sol-gel monolithic materials and their use in a variety of applications. J. Sep. Sci. 2005, 28, 1893–1908. [Google Scholar] [CrossRef]
- Ma, J.; Liang, Z.; Qiao, X.; Deng, Q.; Tao, D.; Zhang, L.; Zhang, Y. Organic-inorganic hybrid silica monolith based immobilized trypsin reactor with high enzymatic activity. Anal. Chem. 2008, 80, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, R.; Wang, F.; Ren, L.; Dong, J.; Liu, Z.; Zou, H. "One-pot" process for fabrication of organic-silica hybrid monolithic capillary columns using organic monomer and alkoxysilane. Anal. Chem. 2009, 81, 3529–3536. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Dai, S.; Guiochon, G. A graphitized-carbon monolithic column. Anal. Chem. 2003, 75, 4904–4912. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Fujikawa, K.; Pornsuriyasak, P.; Alla, A.J.; Ganesh, N.V.; Demchenko, A.V.; Stine, K.J. Lectin-carbohydrate interactions on nanoporous gold monoliths. New J. Chem. 2013, 37, 2150–2165. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.H.; Schallom, J.R.; Ganesh, N.V.; Fujikawa, K.; Demchenko, A.V.; Stine, K.J. Characterization of protein immobilization on nanoporous gold using atomic force microscopy and scanning electron microscopy. Nanoscale 2011, 3, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Sklenářová, H.; Chocholouš, P.; Koblová, P.; Zahálka, L.; Šatínský, D.; Matysová, L.; Solich, P. High-resolution monolithic columns—A new tool for effective and quick separation. Anal. Bioanal. Chem. 2013, 405, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Fekete, S.; Veuthey, J.-L.; Eeltink, S.; Guillarme, D. Comparative study of recent wide-pore materials of different stationary phase morphology, applied for the reversed-phase analysis of recombinant monoclonal antibodies. Anal. Bioanal. Chem. 2013, 405, 3137–3151. [Google Scholar] [CrossRef] [PubMed]
- Namera, A.; Nakamoto, A.; Saito, T.; Miyazaki, S. Monolith as a new sample preparation material: Recent devices and applications. J. Sep. Sci. 2011, 34, 901–924. [Google Scholar] [CrossRef] [PubMed]
- Vlakh, E.G.; Tennikova, T.B. Preparation of methacrylate monoliths. J. Sep. Sci. 2007, 30, 2801–2813. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, D.C. Preparation, structure and morphology of polymer supports. Chem. Commun. 1998, 2275–2286. [Google Scholar] [CrossRef]
- Rieux, L.; Niederländer, H.; Verpoorte, E.; Bischoff, R. Silica monolithic columns: Synthesis, characterisation and applications to the analysis of biological molecules. J. Sep. Sci. 2005, 28, 1628–1641. [Google Scholar] [CrossRef] [PubMed]
- Arrua, R.D.; Alvarez Igarzabal, C.I. Macroporous monolithic supports for affinity chromatography. J. Sep. Sci. 2011, 34, 1974–1987. [Google Scholar] [PubMed]
- Eeltink, S.; Svec, F. Recent advances in the control of morphology and surface chemistry of porous polymer-based monolithic stationary phases and their application in CEC. Electrophoresis 2007, 28, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Svec, F.; Frechet, J.M. New designs of macroporous polymers and supports: From separation to biocatalysis. Science 1996, 273, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Sun, L.; Liang, Z.; Zhang, J.; Wang, H.; Zhang, L.; Zhang, W.; Zhang, Y. Rapid protein digestion and identification using monolithic enzymatic microreactor coupled with nano-liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. A 2006, 1106, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liang, Z.; Qiao, X.; Deng, Q.; Tao, D.; Zhang, L.; Zhang, Y. Organic−inorganic hybrid silica monolith based immobilized trypsin reactor with high enzymatic activity. Anal. Chem. 2008, 80, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Huang, H.; Sun, X.; Lin, Y.; Zhang, L.; Chen, G. Monolithic column based on a poly(glycidyl methacrylate-co-4-vinylphenylboronic acid-co-ethylene dimethacrylate) copolymer for capillary liquid chromatography of small molecules and proteins. J. Chromatogr. A 2012, 1246, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Levkin, P.A.; Eeltink, S.; Stratton, T.R.; Brennen, R.; Robotti, K.; Yin, H.; Killeen, K.; Svec, F.; Fréchet, J.M.J. Monolithic porous polymer stationary phases in polyimide chips for the fast high-performance liquid chromatography separation of proteins and peptides. J. Chromatogr. A 2008, 1200, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Bandari, R.; Kuballa, J.; Buchmeiser, M.R. Ring-opening metathesis polymerization-derived, lectin-functionalized monolithic supports for affinity separation of glycoproteins. J. Sep. Sci. 2013, 36, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bedair, M.; El Rassi, Z. Affinity chromatography with monolithic capillary columns: I. Polymethacrylate monoliths with immobilized mannan for the separation of mannose-binding proteins by capillary electrochromatography and nano-scale liquid chromatography. J. Chromatogr. A 2004, 1044, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Ericson, C.; Holm, J.; Ericson, T.; Hjertén, S. Electroosmosis- and pressure-driven chromatography in chips using continuous beds. Anal. Chem. 1999, 72, 81–87. [Google Scholar] [CrossRef]
- Wu, R.; Hu, L.; Wang, F.; Ye, M.; Zou, H. Recent development of monolithic stationary phases with emphasis on microscale chromatographic separation. J. Chromatogr. A 2008, 1184, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Sondergeld, L.J.; Bush, M.E.; Bellinger, A.; Bushey, M.M. Butyl acrylate porous polymer monoliths in fused-silica capillaries for use in capillary electrochromatography. J. Chromatogr. A 2003, 1004, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Chirica, G.S.; Remcho, V.T. A simple procedure for the preparation of fritless columns by entrapping conventional high performance liquid chromatography sorbents. Electrophoresis 2000, 21, 3093–3101. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, C. Recent developments in column technology for fritless packed column. Chromatography 2001, 22, 145–150. [Google Scholar]
- Bragg, W.; Shamsi, S.A. Development of a fritless packed column for capillary electrochromatography-mass spectrometry. J. Chromatogr. A 2011, 1218, 8691–8700. [Google Scholar] [CrossRef] [PubMed]
- Hahn, R.; Jungbauer, A. Peak broadening in protein chromatography with monoliths at very fast separations. Anal. Chem. 2000, 72, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Bailey, U.M.; Schulz, B.L. Deglycosylation systematically improves N-glycoprotein identification in liquid chromatography-tandem mass spectrometry proteomics for analysis of cell wall stress responses in Saccharomyces cerevisiae lacking Alg3p. J. Chromatogr. B 2013, 923–924, 16–21. [Google Scholar]
- Tarentino, A.L.; Gomez, C.M.; Plummer, T.H. Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry 1985, 24, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Altmann, F.; Marz, L. Peptide-N4-(N-acetyl-beta-glucosaminyl)asparagine amidase F cannot release glycans with fucose attached alpha 1----3 to the asparagine-linked N-acetylglucosamine residue. Eur. J. Biochem. 1991, 199, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Cingoz, A.; Hugon-Chapuis, F.; Pichon, V. Total on-line analysis of a target protein from plasma by immunoextraction, digestion and liquid chromatography-mass spectrometry. J. Chromatogr. B 2010, 878, 213–221. [Google Scholar] [CrossRef]
- Bruyneel, B.; Hoos, J.S.; Smoluch, M.T.; Lingeman, H.; Niessen, W.M.; Irth, H. Trace analysis of proteins using postseparation solution-phase digestion and electrospray mass spectrometric detection of marker peptides. Anal. Chem. 2007, 79, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Kuster, B.; Naven, T.J.; Harvey, D.J. Rapid approach for sequencing neutral oligosaccharides by exoglycosidase digestion and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Mass Spectrom. 1996, 31, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Palm, A.K.; Novotny, M.V. Analytical characterization of a facile porous polymer monolithic trypsin microreactor enabling peptide mass mapping using mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.S.; Rohr, T.; Svec, F.; Frechet, J.M. High-throughput peptide mass mapping using a microdevice containing trypsin immobilized on a porous polymer monolith coupled to MALDI TOF and ESI TOF mass spectrometers. J. Proteome Res. 2002, 1, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.J.; Ferrance, J.; Sanders, J.C.; Landers, J.P. A microchip-based proteolytic digestion system driven by electroosmotic pumping. Lab Chip 2003, 3, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Slysz, G.W.; Schriemer, D.C. On-column digestion of proteins in aqueous-organic solvents. Rapid Commun. Mass Spectrom. 2003, 17, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Strader, M.B.; Tabb, D.L.; Hervey, W.J.; Pan, C.; Hurst, G.B. Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal. Chem. 2006, 78, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.H.; Means, G.E.; Brown, W.D. Stabilization of bovine trypsin by reductive methylation. Biochim. Biophys. Acta 1977, 492, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Hustoft, H.K.; Malerod, H.; Wilson, S.R.; Reubsaet, L.; Lundanes, E.; Greibrokk, T. A critical review of trypsin digestion for LC-MS based proteomics. In Integrative Proteomics; Leung, H.-C.E., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Wang, J.; Zhou, C.; Zhang, W.; Yao, J.; Lu, H.; Dong, Q.; Zhou, H.; Qin, L. An integrative strategy for quantitative analysis of the N-glycoproteome in complex biological samples. Proteome Sci. 2014, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Harvey, D.J. Derivatization of carbohydrates for analysis by chromatography; electrophoresis and mass spectrometry. J. Chromatogr. B 2011, 879, 1196–1225. [Google Scholar] [CrossRef]
- Hase, S.; Ikenaka, T.; Matsushima, Y. Structure analyses of oligosaccharides by tagging of the reducing end sugars with a fluorescent compound. Biochem. Biophys. Res. Commun. 1978, 85, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hase, S.; Hara, S.; Matsushima, Y. Tagging of sugars with a fluorescent compound, 2-aminopyridine. J. Biochem. 1979, 85, 217–220. [Google Scholar] [PubMed]
- Ritamo, I.; Rabina, J.; Natunen, S.; Valmu, L. Nanoscale reversed-phase liquid chromatography-mass spectrometry of permethylated N-glycans. Anal. Bioanal. Chem. 2013, 405, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S. Recent developments in liquid chromatography and capillary electrophoresis for the analysis of glycoprotein glycans. Anal. Sci. 2013, 29, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Demelbauer, U.; Seidl, A.; Friess, W.; Sorgel, F. Reversed-phase liquid-chromatographic mass spectrometric N-glycan analysis of biopharmaceuticals. Anal. Bioanal. Chem. 2013, 405, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, L.A.; Harvey, D.J.; Vouros, P. Reversed-phase ion-pairing liquid chromatography/ion trap mass spectrometry for the analysis of negatively charged, derivatized glycans. Rapid Commun. Mass Spectrom. 2003, 17, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Guile, G.; Rudd, P.; Wing, D.; Dwek, R. HPLC Strategies for Profiling and Sequencing Oligosaccharides. In A Laboratory Guide to Glycoconjugate Analysis; Jackson, P., Gallagher, J., Eds.; Birkhäuser: Basel, Switzerland, 1995; pp. 199–234. [Google Scholar]
- Mechref, Y.; Novotny, M.V. Miniaturized separation techniques in glycomic investigations. J. Chromatogr. B 2006, 841, 65–78. [Google Scholar] [CrossRef]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wu, R.; Dong, J.; Wu, M.; Zhu, Y.; Zou, H. Recent progress of polar stationary phases in CEC and capillary liquid chromatography. Electrophoresis 2009, 30, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P. Stationary and mobile phases in hydrophilic interaction chromatography: A review. Anal. Chim. Acta 2011, 692, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Schlichtherle-Cerny, H.; Affolter, M.; Cerny, C. Hydrophilic interaction liquid chromatography coupled to electrospray mass spectrometry of small polar compounds in food analysis. Anal. Chem. 2003, 75, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Yang, S.H.; Wigginton, J.G.; Simpkins, J.W.; Schug, K.A. Retention behavior of estrogen metabolites on hydrophilic interaction chromatography stationary phases. J. Sep. Sci. 2010, 33, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Quiming, N.S.; Denola, N.L.; Soliev, A.B.; Saito, Y.; Jinno, K. Retention behavior of ginsenosides on a poly(vinyl alcohol)-bonded stationary phase in hydrophilic interaction chromatography. Anal. Bioanal. Chem. 2007, 389, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Khaledi, M.G. Chiral separations by nonaqueous capillary electrophoresis. Anal. Chem. 1996, 68, 3460–3467. [Google Scholar] [CrossRef] [PubMed]
- Kullolli, M.; Hancock, W.S.; Hincapie, M. Preparation of a high-performance multi-lectin affinity chromatography (HP-M-LAC) adsorbent for the analysis of human plasma glycoproteins. J. Sep. Sci. 2008, 31, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Hongsachart, P.; Huang-Liu, R.; Sinchaikul, S.; Pan, F.-M.; Phutrakul, S.; Chuang, Y.-M.; Yu, C.-J.; Chen, S.-T. Glycoproteomic analysis of WGA-bound glycoprotein biomarkers in sera from patients with lung adenocarcinoma. Electrophoresis 2009, 30, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D.; Kornfeld, S. Fractionation of asparagine-linked oligosaccharides by serial lectin-Agarose affinity chromatography. A rapid, sensitive, and specific technique. J. Biol. Chem. 1982, 257, 11235–11240. [Google Scholar] [PubMed]
- Nishikawa, T.; Kajii, S.; Sato, C.; Yasukawa, Z.; Kitajima, K.; Isobe, M. Alpha-C-mannosyltryptophan is not recognized by conventional mannose-binding lectins. Bioorg. Med. Chem. 2004, 12, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Tajiri, M.; Yoshida, S. Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Anal. Chem. 2004, 76, 6560–6565. [Google Scholar] [CrossRef] [PubMed]
- Potter, O.G.; Breadmore, M.C.; Hilder, E.F. Boronate functionalised polymer monoliths for microscale affinity chromatography. Analyst 2006, 131, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Liu, Z.; Dong, M.; Ye, M.; Zou, H. Synthesis and characterization of a new boronate affinity monolithic capillary for specific capture of cis-diol-containing compounds. J. Chromatogr. A 2009, 1216, 4768–4774. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Liu, Y.; Dong, M.; Liu, Z. Synthesis of hydrophilic boronate affinity monolithic capillary for specific capture of glycoproteins by capillary liquid chromatography. J. Chromatogr. A 2009, 1216, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z. Recent advances in monolithic column-based boronate-affinity chromatography. Trends Anal. Chem. 2012, 37, 148–161. [Google Scholar] [CrossRef]
- Otsuka, H.; Uchimura, E.; Koshino, H.; Okano, T.; Kataoka, K. Anomalous binding profile of phenylboronic acid with N-acetylneuraminic acid (Neu5Ac) in aqueous solution with varying pH. J. Am. Chem. Soc. 2003, 125, 3493–3502. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Liu, Z.; Liu, Y.; Dou, P.; Chen, H.Y. Ring-opening polymerization with synergistic co-monomers: access to a boronate-functionalized polymeric monolith for the specific capture of cis-diol-containing biomolecules under neutral conditions. Angew. Chem. Int. Ed. Engl. 2009, 48, 6704–6707. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; James, S.L. A metal-organic gel used as a template for a porous organic polymer. Chem. Commun. 2005, 1555–1556. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, A.; Yates, J.R. Multidimensional LC separations in shotgun proteomics. Anal. Chem. 2008, 80, 7187–7193. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Miliotis, T.; Marko-Varga, G.; Bischoff, R.; Unger, K.K. An automated on-line multidimensional HPLC system for protein and peptide mapping with integrated sample preparation. Anal. Chem. 2002, 74, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Stange, D.; van de Wetering, M.; Clevers, H.; Heck, A.J.R.; Mohammed, S. Highly sensitive proteome analysis of FACS-sorted adult colon stem cells. J. Proteome Res. 2011, 10, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.M.; Diamond, D.L.; Chan, E.Y.; Gritsenko, M.A.; Qian, W.; Stastna, M.; Baas, T.; Camp, D.G., 2nd; Carithers, R.L., Jr.; Smith, R.D.; et al. Proteome analysis of liver cells expressing a full-length hepatitis C virus (HCV) replicon and biopsy specimens of posttransplantation liver from HCV-infected patients. J. Virol. 2005, 79, 7558–7569. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Nakano, T.; Kawamura, T.; Usui, F.; Bando, Y.; Wang, R.; Nishimura, T. Multidimensional protein profiling technology and its application to human plasma proteome. J. Proteome Res. 2004, 3, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Schafer, H.; Klaus, S.; Bunse, C.; Swart, R.; Meyer, H.E. A new fast method for nanoLC-MALDI-TOF/TOF-MS analysis using monolithic columns for peptide preconcentration and separation in proteomic studies. J. Proteome Res. 2007, 6, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Shiyan, S.D.; Bovin, N.V. Carbohydrate composition and immunomodulatory activity of different glycoforms of alpha1-acid glycoprotein. Glycoconjug. J. 1997, 14, 631–638. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alla, A.J.; Stine, K.J. Development of Monolithic Column Materials for the Separation and Analysis of Glycans. Chromatography 2015, 2, 20-65. https://doi.org/10.3390/chromatography2010020
Alla AJ, Stine KJ. Development of Monolithic Column Materials for the Separation and Analysis of Glycans. Chromatography. 2015; 2(1):20-65. https://doi.org/10.3390/chromatography2010020
Chicago/Turabian StyleAlla, Allan J., and Keith J. Stine. 2015. "Development of Monolithic Column Materials for the Separation and Analysis of Glycans" Chromatography 2, no. 1: 20-65. https://doi.org/10.3390/chromatography2010020
APA StyleAlla, A. J., & Stine, K. J. (2015). Development of Monolithic Column Materials for the Separation and Analysis of Glycans. Chromatography, 2(1), 20-65. https://doi.org/10.3390/chromatography2010020