Procyanidin B2 Protects Neurons from Oxidative, Nitrosative, and Excitotoxic Stress


Abstract
:1. Introduction
2. Materials and Methods
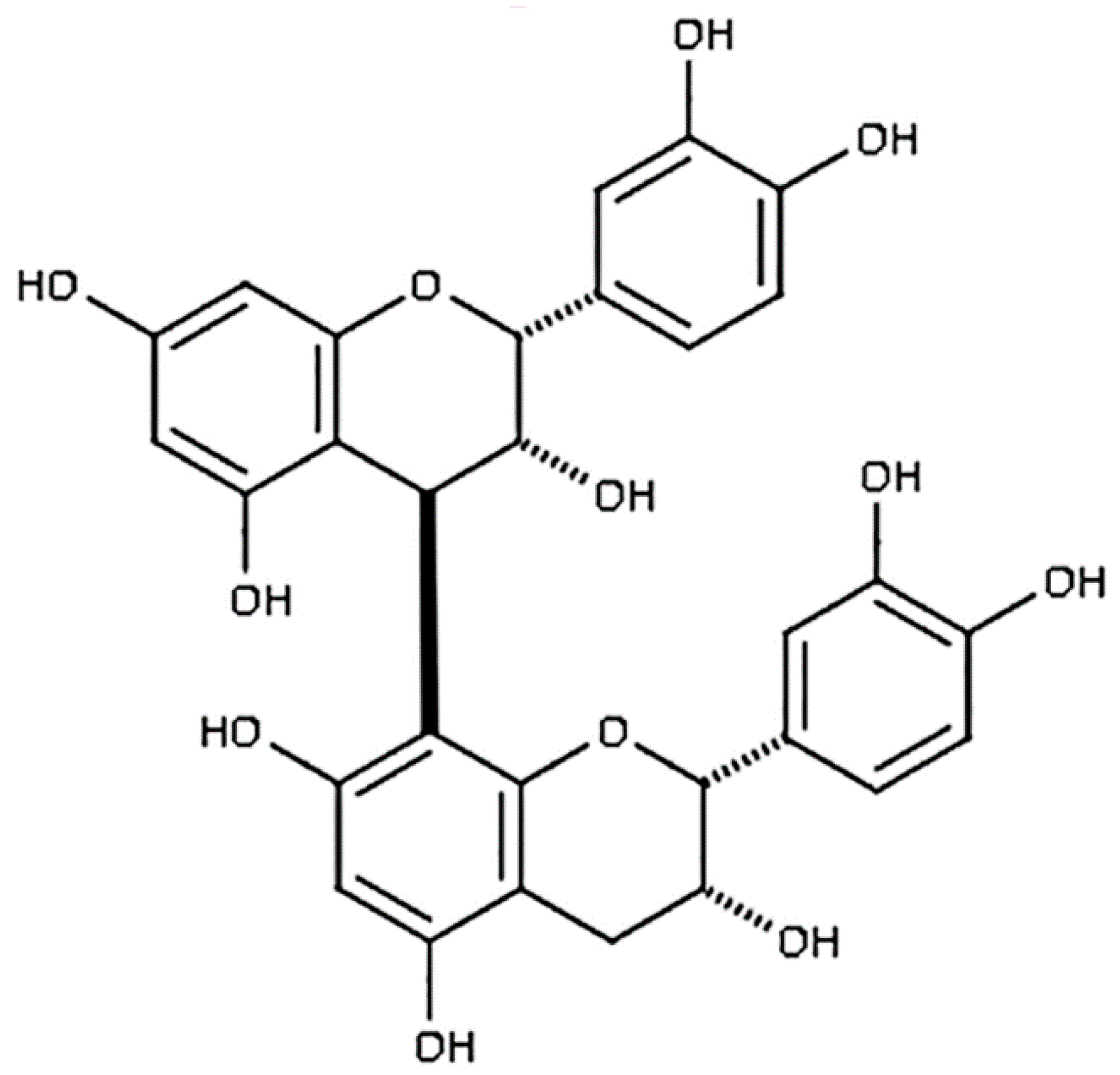
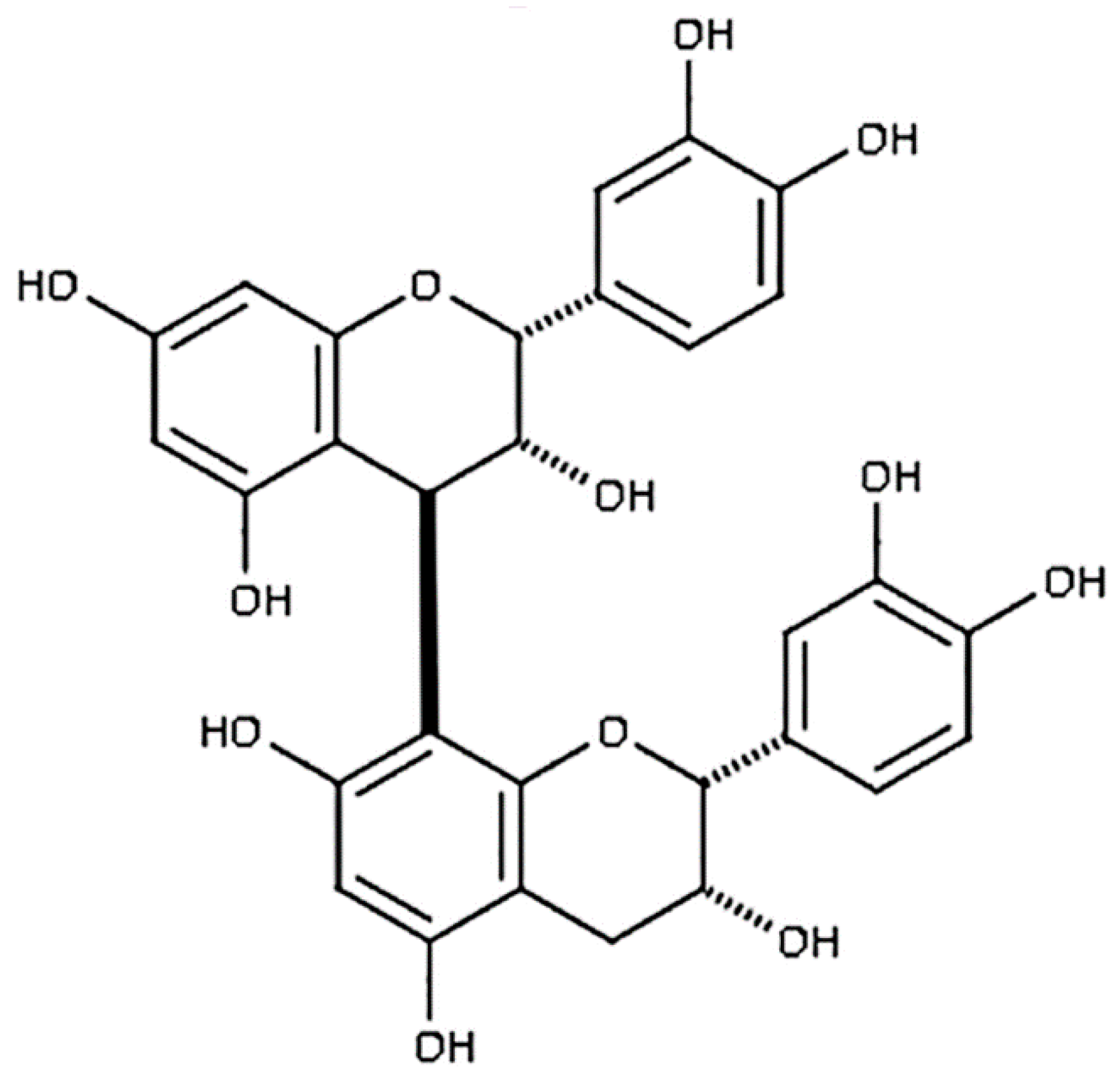
2.1. Chemicals and Reagents
2.2. Cell Culture: Rat Cerebellar Granule Neurons
2.3. Protocol for Treatment with Procyanidin B2
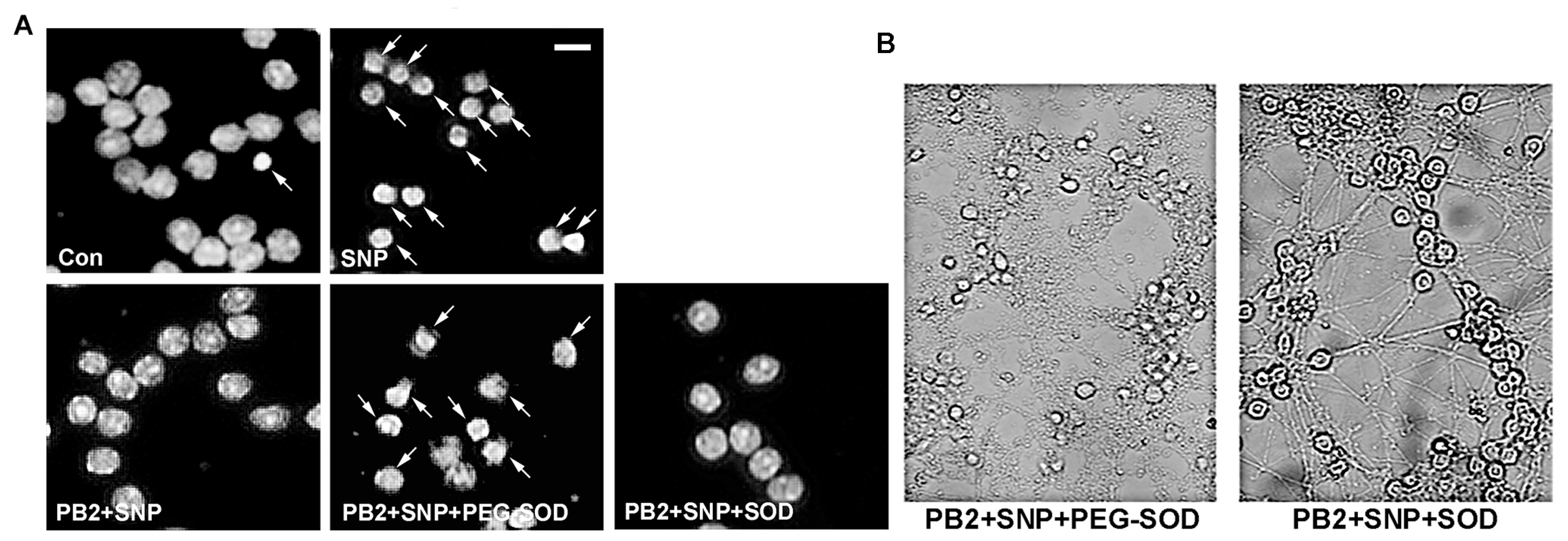
2.4. Protocol for Treatment with PEG-SOD and SOD
2.5. Immunocytochemistry
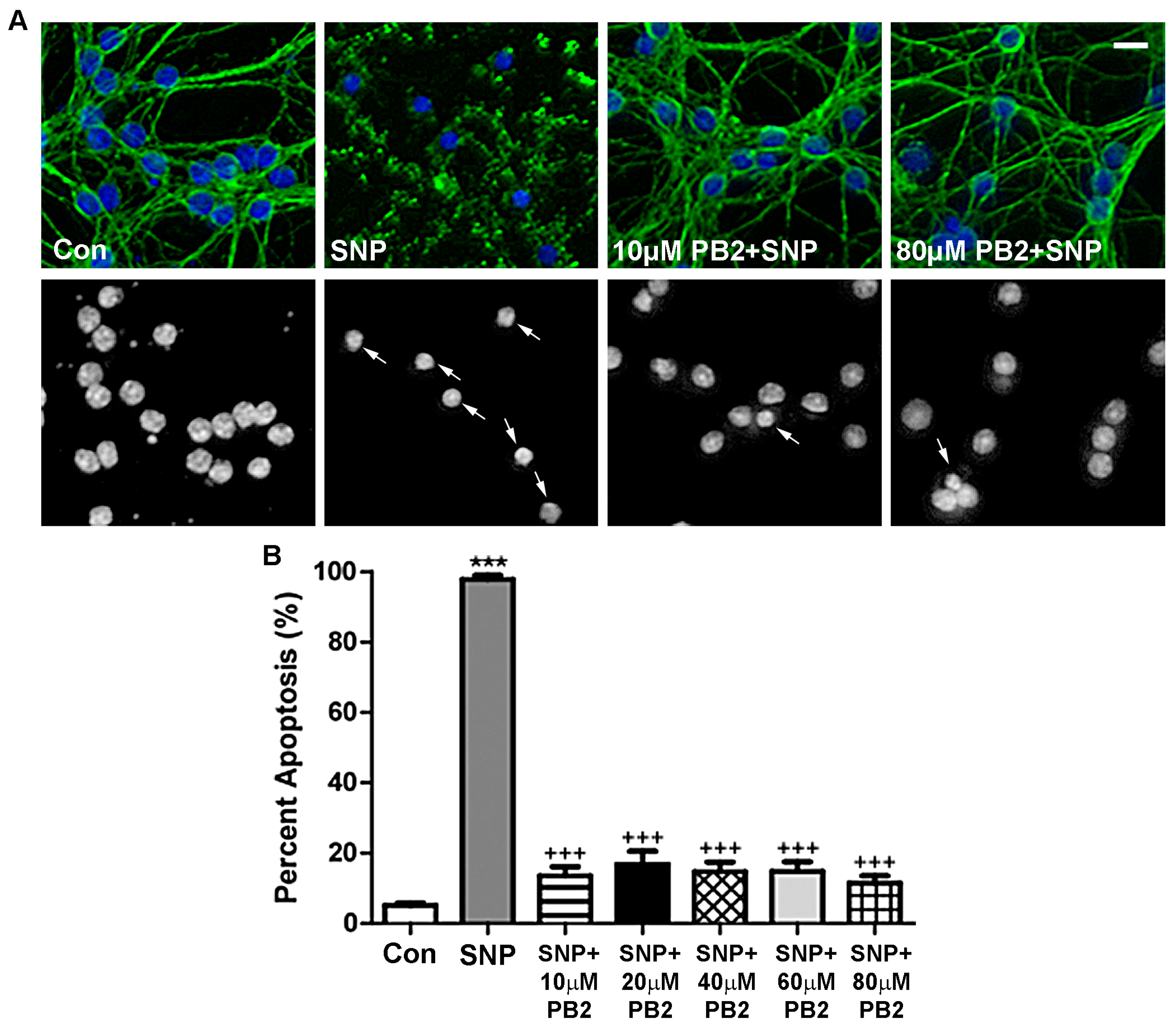
2.6. Analysis of Apoptosis
2.7. MTT Viability Assay
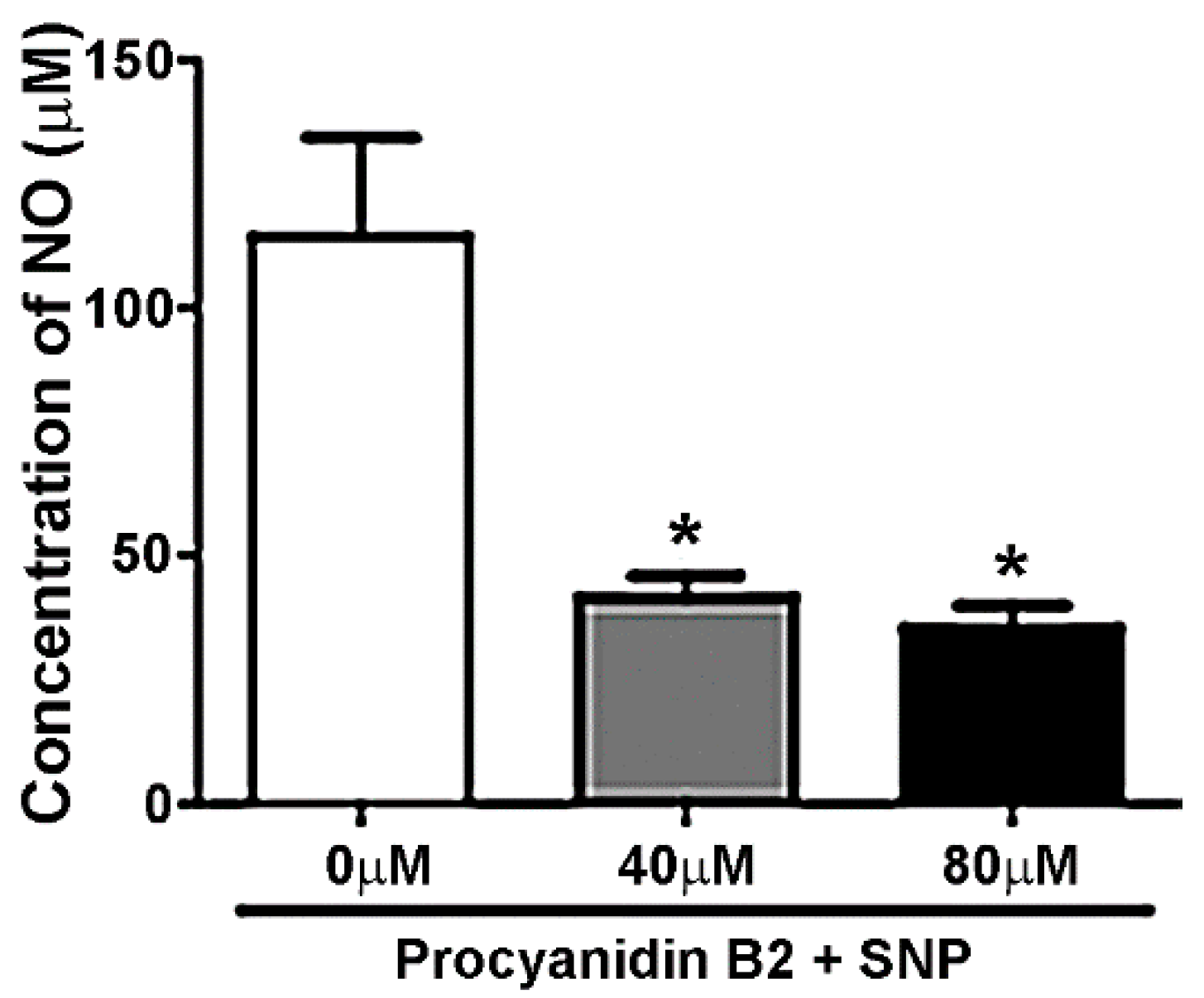
2.8. Nitric Oxide Assay
2.9. Data Analysis
3. Results
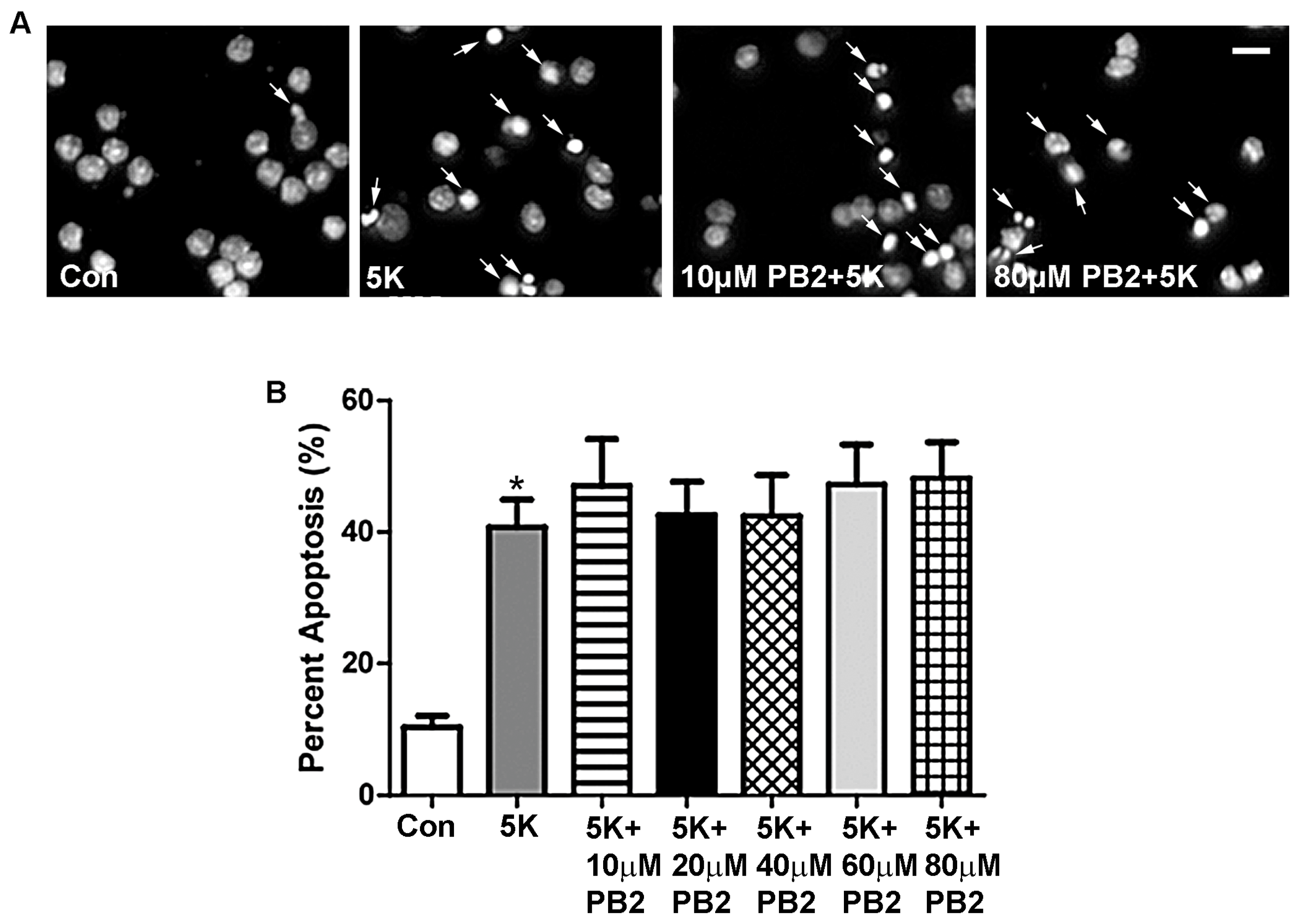
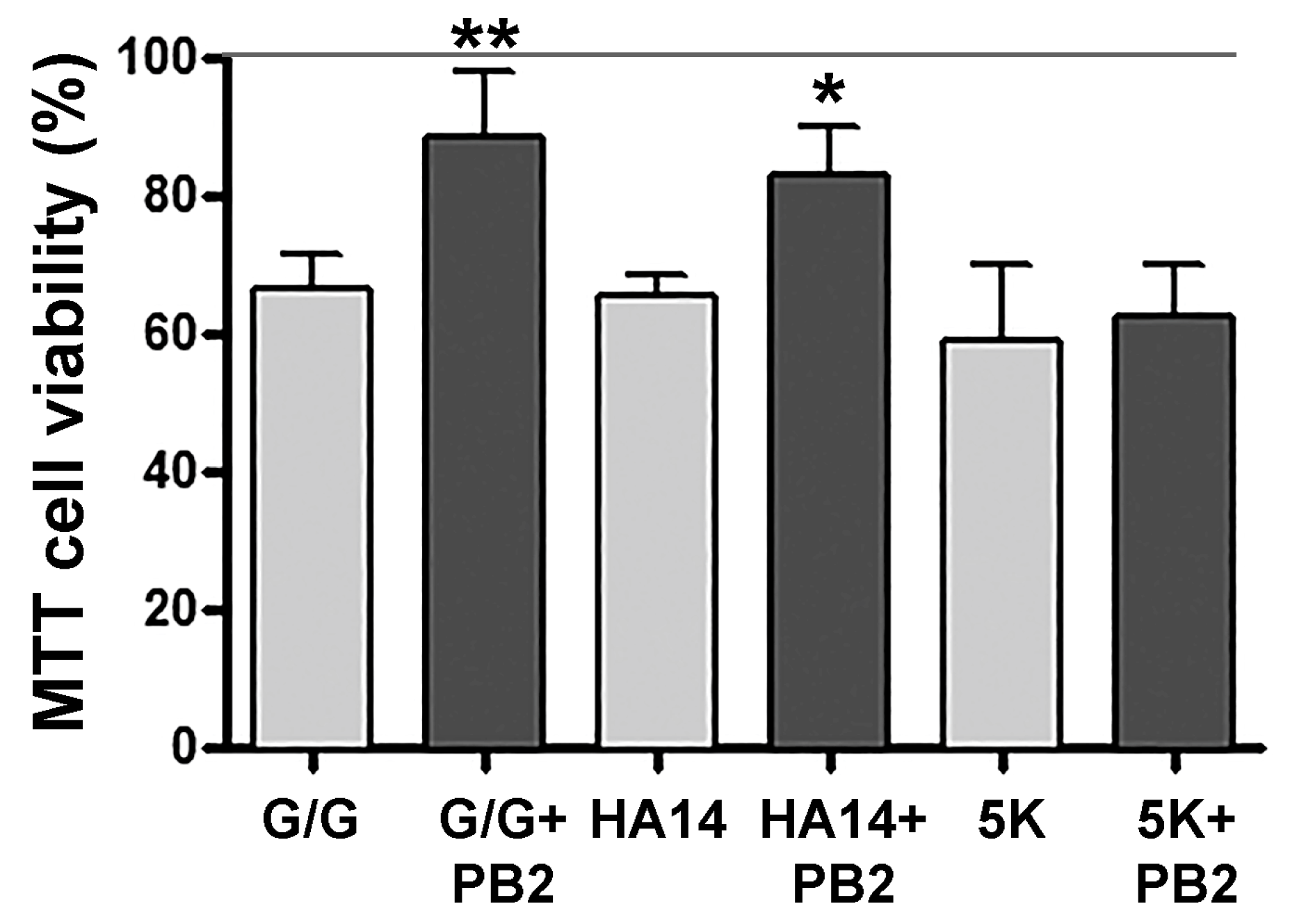
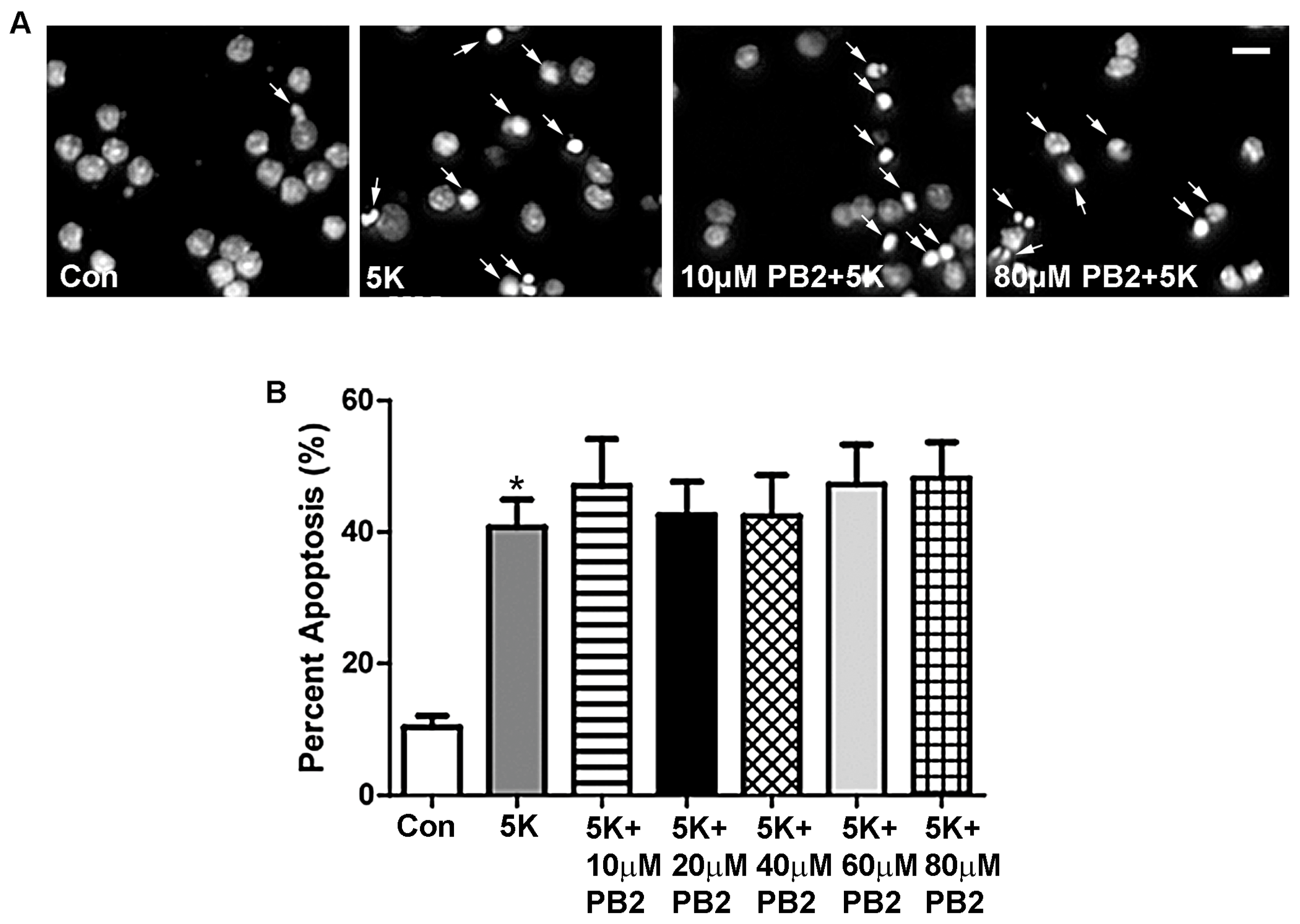
3.1. Procyanidin B2 Does Not Protect against 5K-Induced Apoptosis
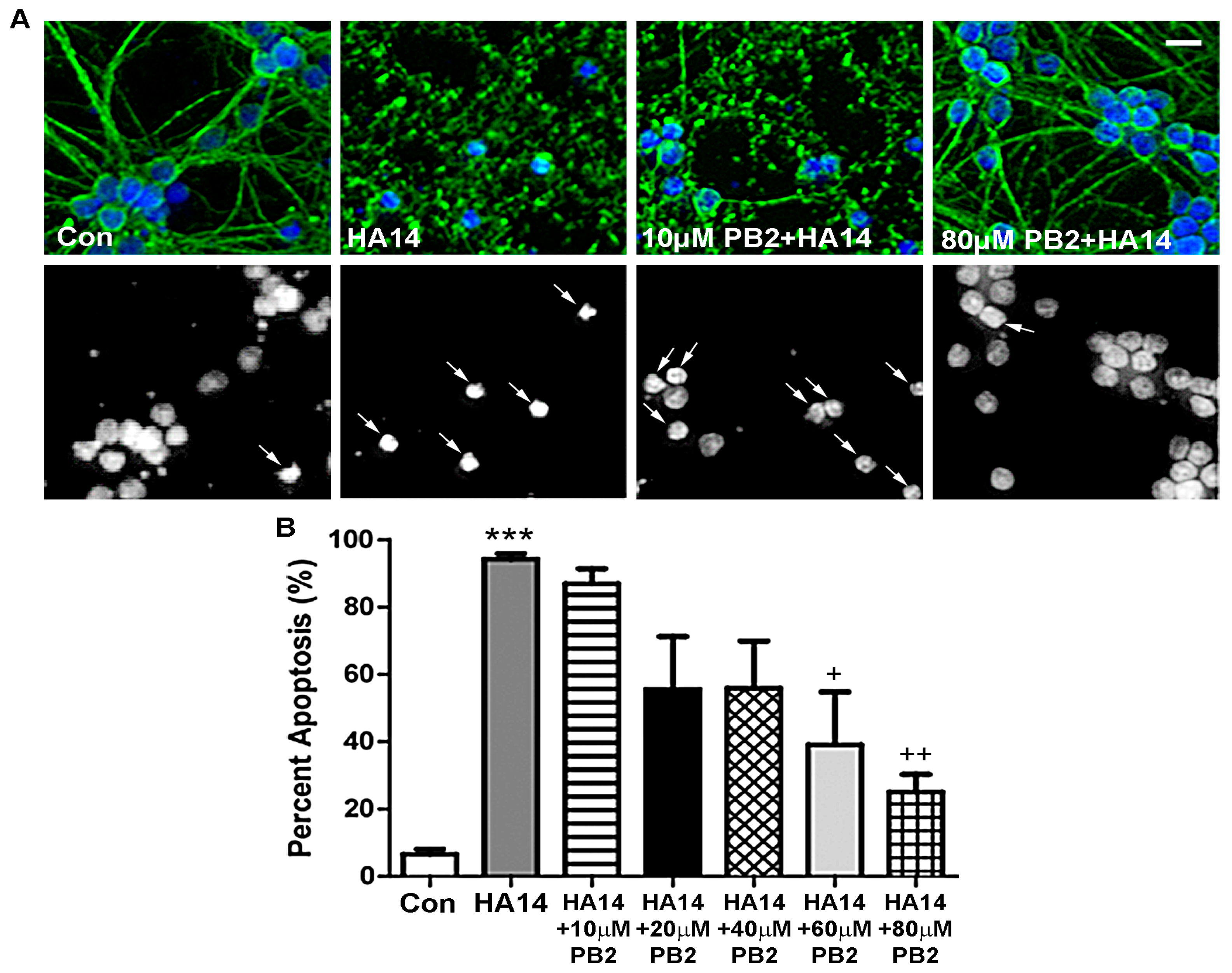
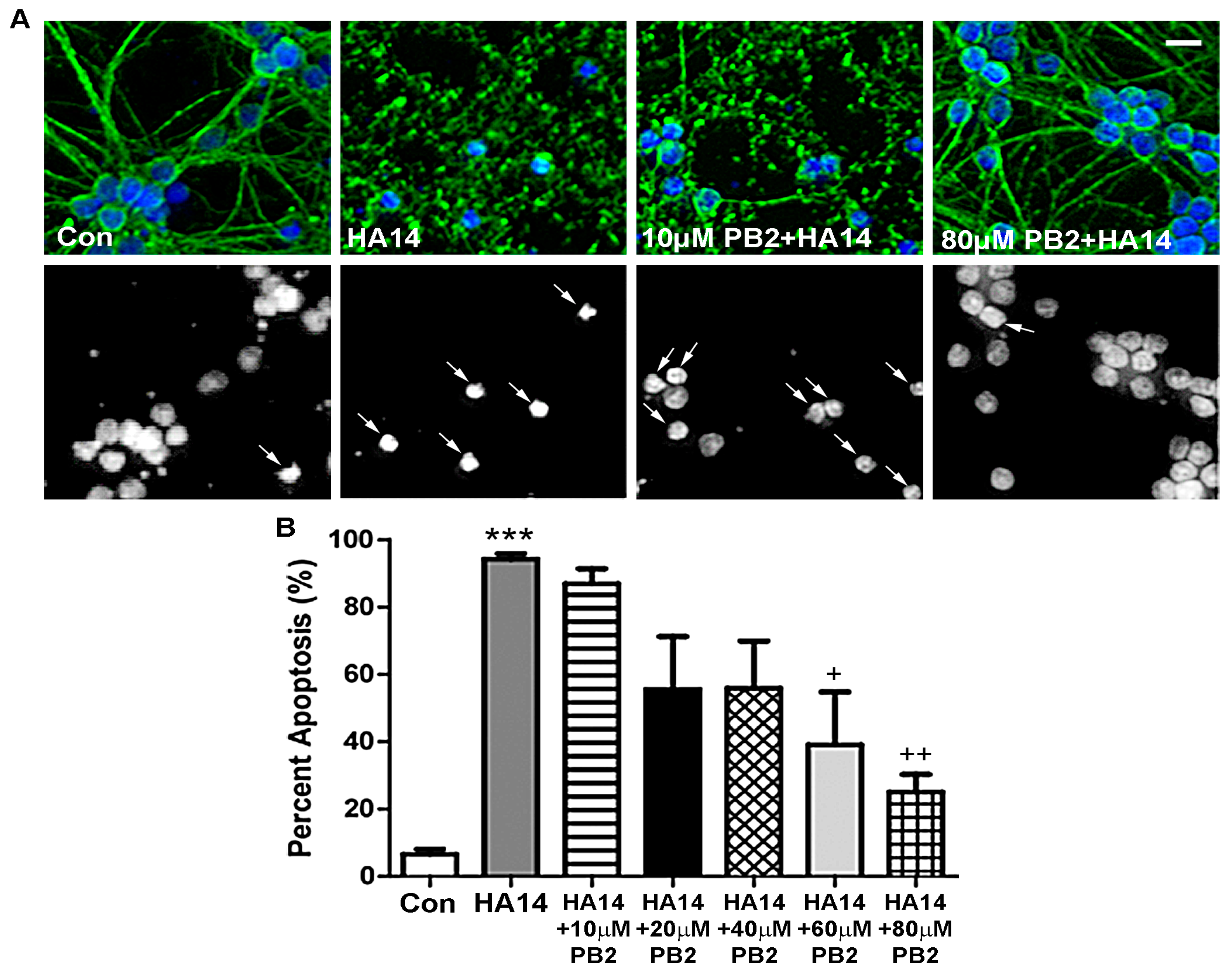
3.2. Procyanidin B2 Protects against Apoptosis Induced by the Bcl-2 Inhibitor HA14-1
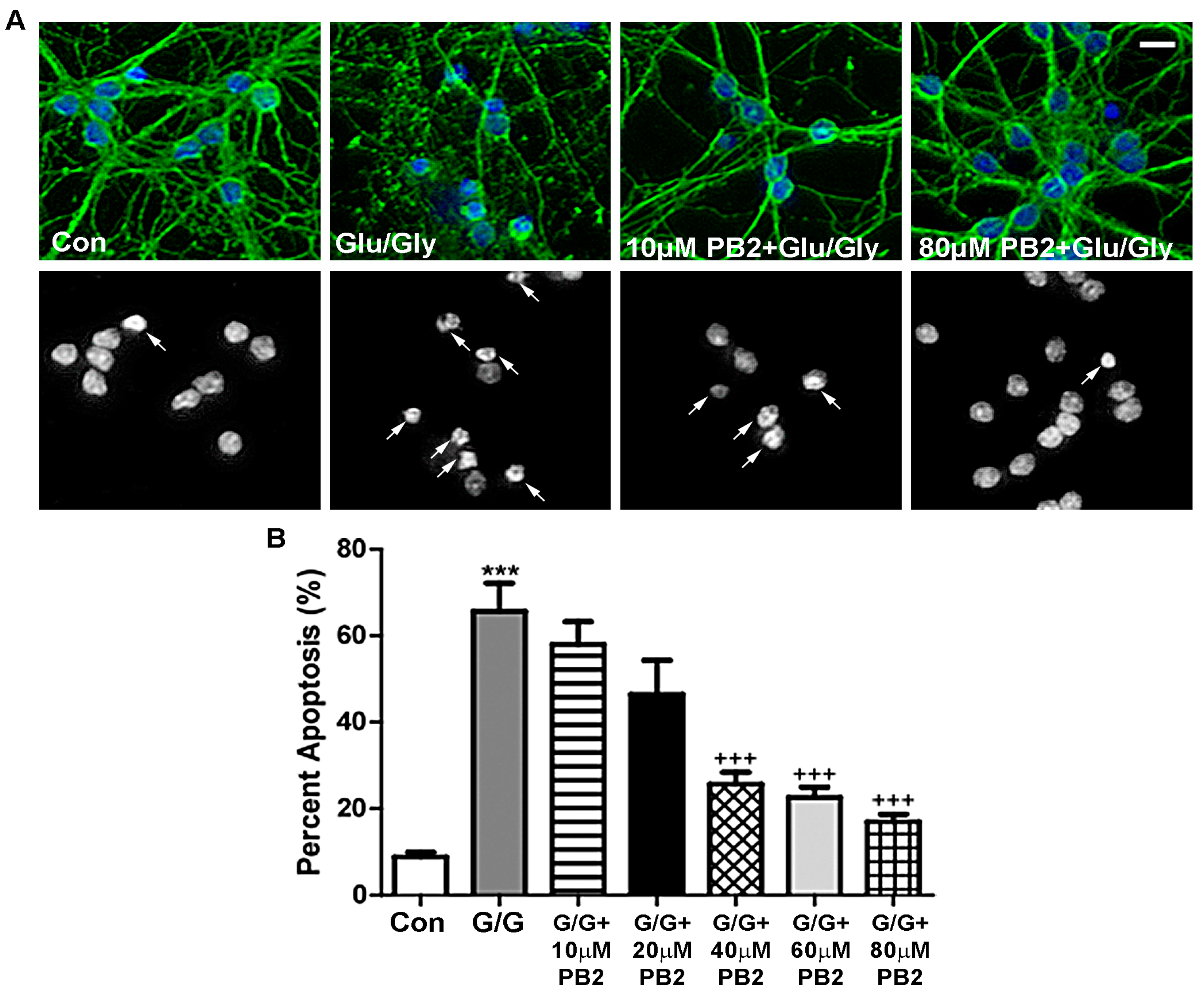
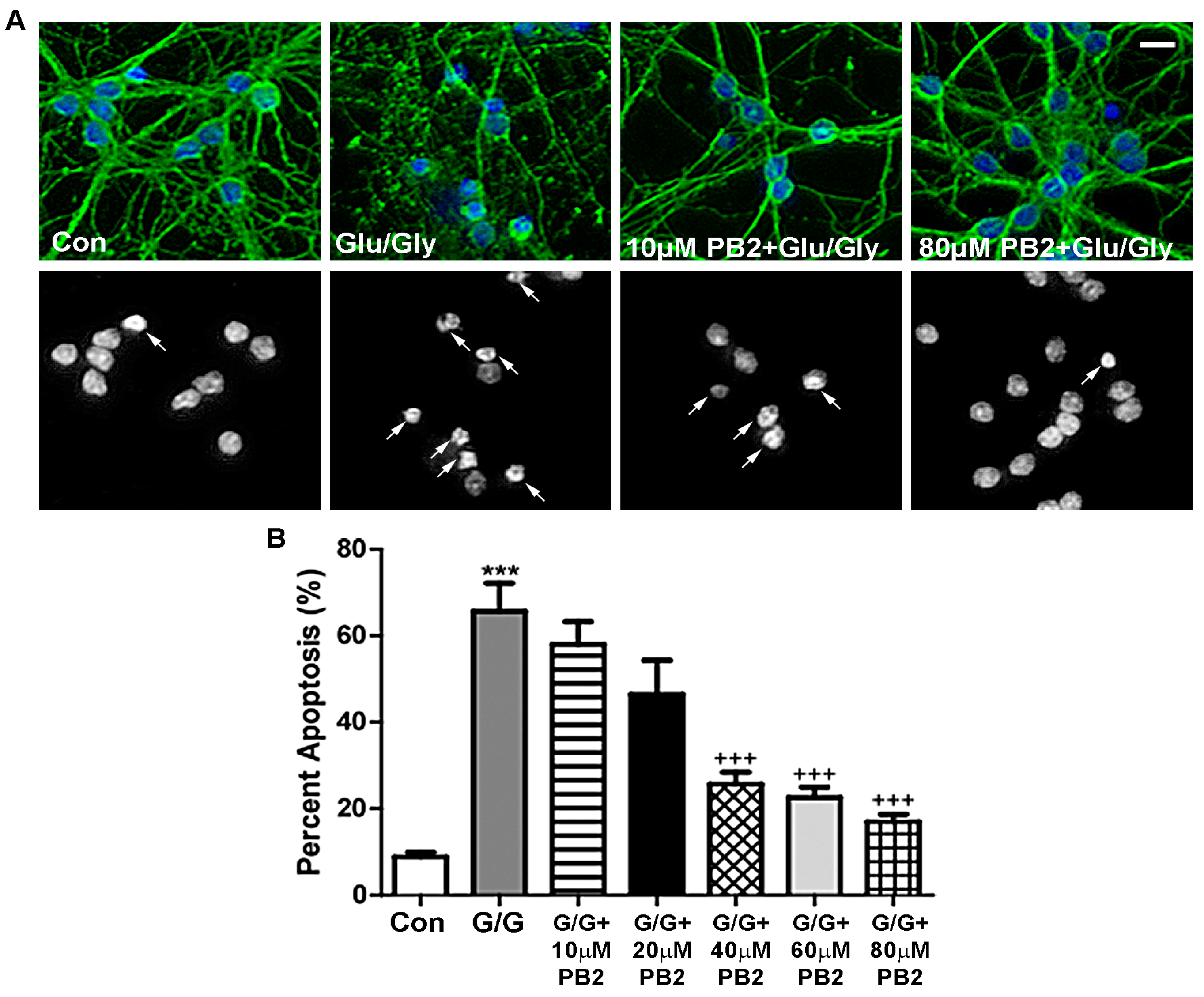
3.3. Procyanidin B2 Protects CGNs from Glutamate-Induced Excitotoxicity
3.4. Evaulation of Procyanidin B2 Neuroprotective Effects by MTT Viability Assay
3.5. Procyanidin B2 Protects CGNs against SNP-Induced Nitrosative Stress
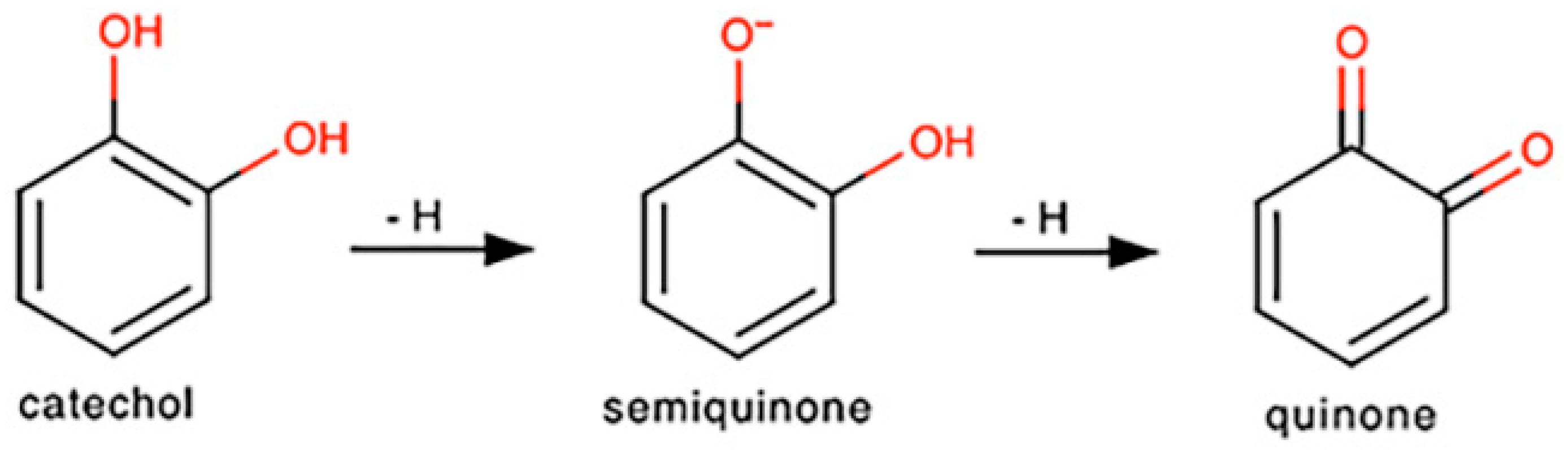
3.6. Procyanidin B2 Reduces Nitric Oxide Concentration in a Cell-Free System
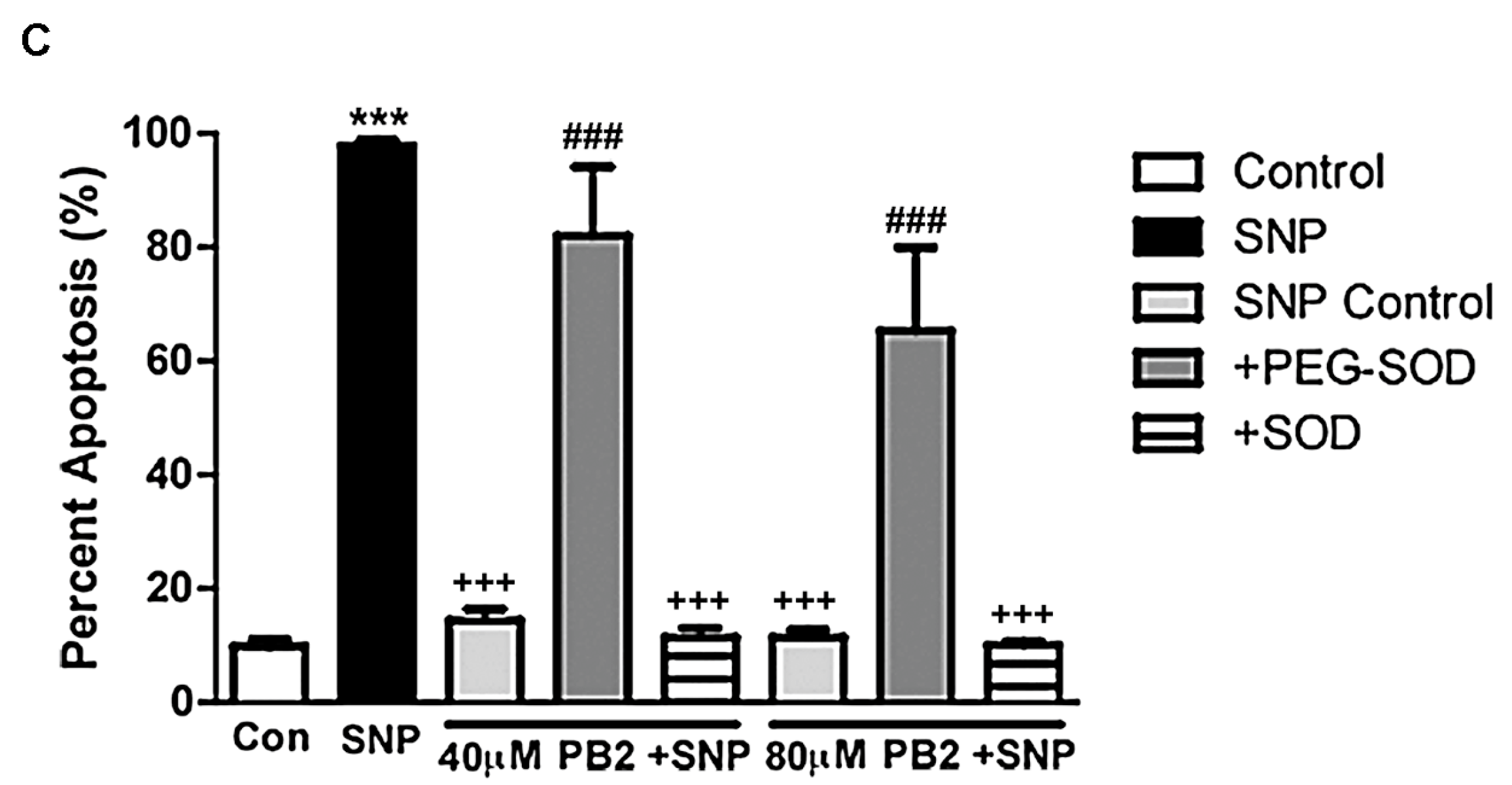
3.7. PEG-SOD Prevents PB2 from Protecting CGNs against SNP-Induced Toxicity
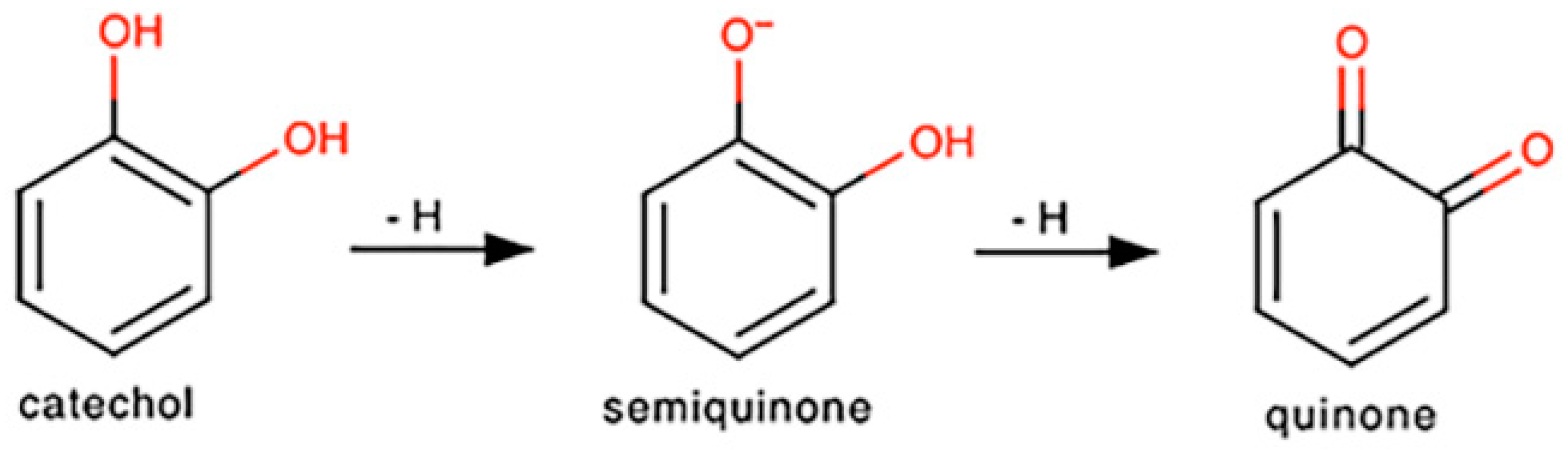
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| CGNs | cerebellar granule neurons |
| GSH | glutathione |
| H2O2 | hydrogen peroxide |
| HA14-1 | 2-amino-6-bromo-α-cyano-3-(ethoxycarbonyl)-4H-1-benzopyran-4-acetic acid ethyl ester |
| iNOS | inducible nitric oxide synthase |
| MOS | mitochondrial oxidative stress; nNOS, neuronal nitric oxide synthase |
| NO• | nitric oxide |
| O2•− | superoxide anion |
| •OH | hydroxyl radical |
| ONOO− | peroxynitrite |
| PB2 | procyanidin B2 |
| PBS | phosphate buffered saline |
| PD | Parkinson’s disease |
| PEG-SOD | polyethylene glycol conjugated superoxide dismutase |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SNP | sodium nitroprusside |
| SOD | superoxide dismutase |
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.A.; Estéves, A.G.; Barbeito, L.; Beckman, J.S. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 2012, 22, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42, S125–S152. [Google Scholar] [PubMed]
- Simoncini, C.; Orsucci, D.; Caldarazzo Ienco, E.; Siciliano, G.; Bonuccelli, U.; Mancuso, M. Alzheimer’s pathogenesis and its link to the mitochondrion. Oxid. Med. Cell. Longev. 2015, 2015, 803942. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic Parkinsonism. Neurobiol. Dis. 2012, 51, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Wu, S.; Yue, Y.; Li, J.; Li, Z.; Li, X.; Niu, Y.; Xiang, J.; Ding, H. Procyanidin B2 attenuates neurological deficits and blood-brain barrier disruption in a rat model of cerebral ischemia. Mol. Nutr. Food Res. 2015, 59, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Leverenz, J.B.; Lau, R.C.; Polissar, N.L.; Neradilek, M.B.; Samii, A.; Standish, L.J. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Mov. Disord. 2015, 30, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Asiimwe, N.; Yeo, S.G.; Kim, M.S.; Jung, J.; Jeong, N.Y. Nitric oxide: Exploring the contextual link with Alzheimer’s disease. Oxid. Med. Cell. Longev. 2016, 2016, 7205747. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Dawson, T.M.; Dawson, V.L. Nitric oxide, S-nitrosylation and neurodegeneration. Cell. Mol. Biol. (Noisy-le-Grand) 2005, 51, 247. [Google Scholar]
- Uehara, T. Accumulation of misfolded protein through nitrosative stress linked to neurodegenerative disorders. Antioxid. Redox Signal. 2007, 9, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Antioxid. Redox Signal. 2011, 14, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.E.; Harris, M. S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions. Front. Chem. 2015, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; David, K.K. Emerging roles of nitric oxide in neurodegeneration. Nitric Oxide 2010, 22, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.P.; Garcia-Nogales, P.; Almeida, A. Provoking neuroprotection by peroxynitrite. Curr. Pharm. Des. 2004, 10, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Miranda, M.; Narayan, M. Nitrosative stress-induced Parkinsonian Lewy-like aggregates prevented through polyphenolic phytochemical analog intervention. Biochem. Biophys. Res. Commun. 2011, 404, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Calviello, G. Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effects of n-3 PUFA in Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Lee, S.R.; Im, K.J.; Suh, S.I.; Jung, J.G. Protective effect of green tea polyphenol (−)-epigallocatechin gallate and other antioxidants on lipid peroxidation in gerbil brain homogenates. Phytother. Res. 2003, 17, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.J.; Lee, C.Y. Strawberry and its anthocyanins reduce oxidative stress-induced apoptosis in PC12 cells. J. Agric. Food Chem. 2005, 53, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbinson, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective anti-apoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.; Hulick, W.; Winter, A.; Ross, E.; Linseman, D. Neuroprotective effects of anthocyanins on apoptosis induced by mitochondrial oxidative stress. Nutr. Neurosci. 2011, 14, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Srividhya, R.; Kalaiselvi, P. Neuroprotective potential of epigallo catechin-3-gallate in PC-12 cells. Neurochem. Res. 2013, 38, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.N.; Ross, E.K.; Khatter, S.; Miller, K.; Linseman, D.A. Chemical basis for the disparate neuropotective effects of the anthocyanins, callistephin and kuromanin, against nitrosative stress. Free Radic. Biol. Med. 2017, 103, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Park, C.S.; Kim, D.J.; Cho, M.H.; Jin, B.K.; Pie, J.E.; Chung, W.G. Prevention of nitric oxide-mediated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease in mice by tea phenolic epigallocatechin 3-gallate. Neurotoxicology 2002, 23, 367–374. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P. Berry fruits for cancer prevention: Current status and future prospects. J. Agric. Food Chem. 2008, 56, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Hecht, S.S.; Carmella, S.G.; Yu, N.; Larue, B.; Henry, C.; McIntyre, C.; Rocha, C.; Lechner, J.F.; Stoner, G.D. Anthocyanins in black raspberries prevent esophageal tumors in rats. Cancer Prev. Res. 2009, 2, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sakano, K.; Mizutani, M.; Murata, M.; Oikawa, S.; Hiraku, Y.; Kawanishi, S. Procyanidin B2 has anti-and pro-oxidant effects on metal-mediated DNA damage. Free Radic. Biol. Med. 2005, 39, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Panickar, K.S.; Cao, H.; Qin, B.; Anderson, R.A. Molecular targets and health benefits of cinnamon. In Molecular Targets and Therapeutic Uses of Spices; World Scientific Publishing Co.: Hackensack, NJ, USA, 2009; pp. 87–116. [Google Scholar]
- Cho, E.S.; Lee, K.W.; Lee, H.J. Cocoa procyanidins protect PC12 cells from hydrogen-peroxide-induced apoptosis by inhibiting activation of p38 MAPK and JNK. Mutat. Res. Fund. Mol. Mech. Mut. 2008, 640, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Jang, Y.J.; Kang, N.J.; Hwang, M.K.; Kim, Y.T.; Lee, K.W.; Lee, H.J. Cocoa procyanidins attenuate 4-hydroxynonenal-induced apoptosis of PC12 cells by directly inhibiting mitogen-activated protein kinase kinase 4 activity. Free Radic. Biol. Med. 2009, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Laessig, T.; Meintzer, M.K.; McClure, M.; Barth, H.; Aktories, K.; Heidenreich, K.A. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J. Biol. Chem. 2001, 276, 39123–39131. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989–10993. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Phelps, R.A.; Bouchard, R.J.; Le, S.S.; Laessig, T.A.; McClure, M.L.; Heidenreich, K.A. Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J. Neurosci. 2002, 22, 9287–9297. [Google Scholar] [PubMed]
- Wang, J.; Liu, D.; Zhang, Z.; Shan, S.; Han, X. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.K.; Loucks, F.A.; Schroeder, E.K.; Bouchard, R.J.; Tyler, K.L.; Linseman, D.A. Glutathione binding to the Bcl-2 homology-3 domain groove: A molecular basis for Bcl-2 antioxidant function at mitochondria. J. Biol. Chem. 2007, 282, 29296–29304. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Marquardt, K.; Lash, L.H.; Linseman, D.A. Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Radic. Biol. Med. 2012, 52, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.M.; Bristow, D.R. N-methyl-d-aspartate receptor desensitisation is neuroprotective by inhibiting glutamate-induced apoptotic-like death. J. Neurochem. 1998, 70, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H. NO at work. Cell 1994, 78, 919–925. [Google Scholar] [CrossRef]
- Huie, R.E.; Padmaja, S. The reaction of NO with superoxide. Free Radic. Res. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Meunier, S.; Hanédanian, M.; Murr, D.E.; Nowaczyk, S.; Le Gall, T.; Pin, S.; Renault, J.; Bouquet, D.; Creminon, C.; Mioskowski, C.; et al. High-throughput evaluation of antioxidant and pro-oxidant activities of polyphenols with thymidine protection assays. ChemBioChem 2005, 6, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Galinanes, M.; Qiu, Y.; Ezrin, A.; Hearse, D.J. PEG-SOD and myocardial protection. Studies in the blood-and crystalloid-perfused rabbit and rat hearts. Circulation 1992, 86, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W. Oxidative stress in amyotrophic lateral sclerosis. J. Neurol. 2000, 247, I1–I6. [Google Scholar] [CrossRef] [PubMed]
- Edens, B.M.; Miller, N.; Ma, Y.C. Impaired autophagy and defective mitochondrial function: Converging paths on the road to motor neuron degeneration. Front. Cell. Neurosci. 2016, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Goto, H.; Kogure, T.; Shibahara, N.; Sakakibara, I.; Sasaki, H.; Terasawa, K. Protective effect of phenolic compounds isolated from the hooks and stems of Uncaria sinensis on glutamate-induced neuronal death. Am. J. Chin. Med. 2001, 29, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, 233S–241S. [Google Scholar] [CrossRef]
- Heath, P.R.; Shaw, P.J. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 438–458. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural Transm. (Vienna) 2014, 121, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Dulebohn, R.V.; Yi, W.; Srivastava, A.; Akoh, C.C.; Krewer, G.; Fischer, J.G. Effects of blueberry (Vaccinium ashei) on DNA damage, lipid peroxidation, and phase II enzyme activities in rats. J. Agric. Food Chem. 2008, 56, 11700–11706. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Ni, H.M.; Wang, S.Y.; Tourkova, I.L.; Shurin, M.R.; Harada, H.; Yin, X.M. Cyanidin-3-rutinoside, a natural polyphenol antioxidant, selectively kills leukemic cells by induction of oxidative stress. J. Biol. Chem. 2007, 282, 13468–13476. [Google Scholar] [CrossRef] [PubMed]
- Chichirau, A.; Flueraru, M.; Chepelev, L.L.; Wright, J.S.; Willmore, W.G.; Durst, T.; Hussain, H.H.; Charron, M. Mechanism of cytotoxicity of catechols and a naphthalenediol in PC12-AC cells: The connection between extracellular autoxidation and molecular electronic structure. Free Radic. Biol. Med. 2005, 38, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.H.; Babic, G.; Durst, T.; Wright, J.S.; Flueraru, M.; Chichirau, A.; Chepelev, L.L. Development of novel antioxidants: Design, synthesis, and reactivity. J. Org. Chem. 2003, 68, 7023–7032. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Nogales, P.; Almeida, A.; Bolaños, J.P. Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 2003, 278, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Osakabe, N.; Natsume, M.; Terao, J. Absorption and urinary excretion of procyanidin B2 [epicatechin-(4β-8)-epicatechin] in rats. Free Radic. Biol. Med. 2002, 33, 142–148. [Google Scholar] [CrossRef]
- Rios, L.Y.; Bennett, R.N.; Lazarus, S.A.; Rémésy, C.; Scalbert, A.; Williamson, G. Cocoa procyanidins are stable during gastric transit in humans. Am. J. Clin. Nutr. 2002, 76, 1106–1110. [Google Scholar] [PubMed]
- Holt, R.R.; Lazarus, S.A.; Sullards, M.C.; Zhu, Q.Y.; Schramm, D.D.; Hammerstone, J.F.; Fraga, C.G.; Schmitz, H.H.; Keen, C.L. Procyanidin dimer B2 [epicatechin-(4β-8)-epicatechin] in human plasma after the consumption of a flavanol-rich cocoa. Am. J. Clin. Nutr. 2002, 76, 798–804. [Google Scholar] [PubMed]
- Serra, A.; Macià, A.; Romero, M.P.; Valls, J.; Bladé, C.; Arola, L.; Motilva, M.J. Bioavailability of procyanidin dimers and trimers and matrix food effects in in vitro and in vivo models. Br. J. Nutr. 2010, 103, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Macià, A.; Rubió, L.; Anglès, N.; Ortega, N.; Morelló, J.R.; Romero, M.; Motilva, M.J. Distribution of procyanidins and their metabolites in rat plasma and tissues in relation to ingestion of procyanidin-enriched or procyanidin-rich cocoa creams. Eur. J. Nutr. 2012, 52, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Yamakoshi, J.; Saito, M.; Kataoka, S.; Kikuchi, M. Safety evaluation of proanthocyanidin-rich extract from grape seeds. Food Chem. Toxicol. 2002, 40, 599–607. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutcliffe, T.C.; Winter, A.N.; Punessen, N.C.; Linseman, D.A. Procyanidin B2 Protects Neurons from Oxidative, Nitrosative, and Excitotoxic Stress. Antioxidants 2017, 6, 77. https://doi.org/10.3390/antiox6040077
Sutcliffe TC, Winter AN, Punessen NC, Linseman DA. Procyanidin B2 Protects Neurons from Oxidative, Nitrosative, and Excitotoxic Stress. Antioxidants. 2017; 6(4):77. https://doi.org/10.3390/antiox6040077
Chicago/Turabian StyleSutcliffe, Taylor C., Aimee N. Winter, Noelle C. Punessen, and Daniel A. Linseman. 2017. "Procyanidin B2 Protects Neurons from Oxidative, Nitrosative, and Excitotoxic Stress" Antioxidants 6, no. 4: 77. https://doi.org/10.3390/antiox6040077
APA StyleSutcliffe, T. C., Winter, A. N., Punessen, N. C., & Linseman, D. A. (2017). Procyanidin B2 Protects Neurons from Oxidative, Nitrosative, and Excitotoxic Stress. Antioxidants, 6(4), 77. https://doi.org/10.3390/antiox6040077