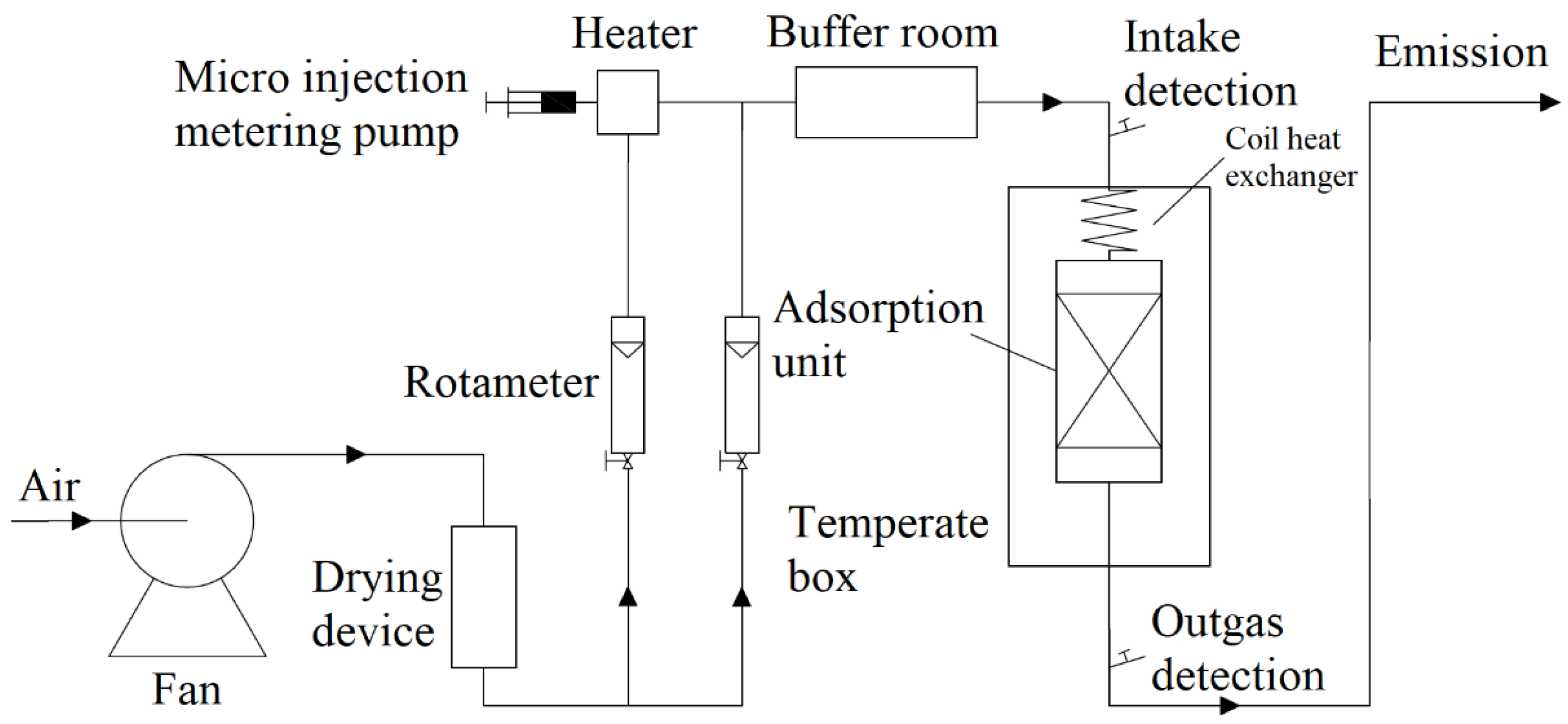
3.1. Adsorption Equilibrium Isotherm
The foundation for our subsequent research was the isotherm of adsorption of toluene on activated carbon. We weighed the activated carbon bed before adsorption. At a particular temperature and concentration, the toluene gas passed through the activated carbon. We used a Smart FID to monitor the concentration of toluene gas prior to and following the stainless steel adsorber. We weighed the saturated activated carbon after the inlet concentration was stable and equal to the outlet concentration. The outlet concentration was used to draw the adsorption breakthrough curve.
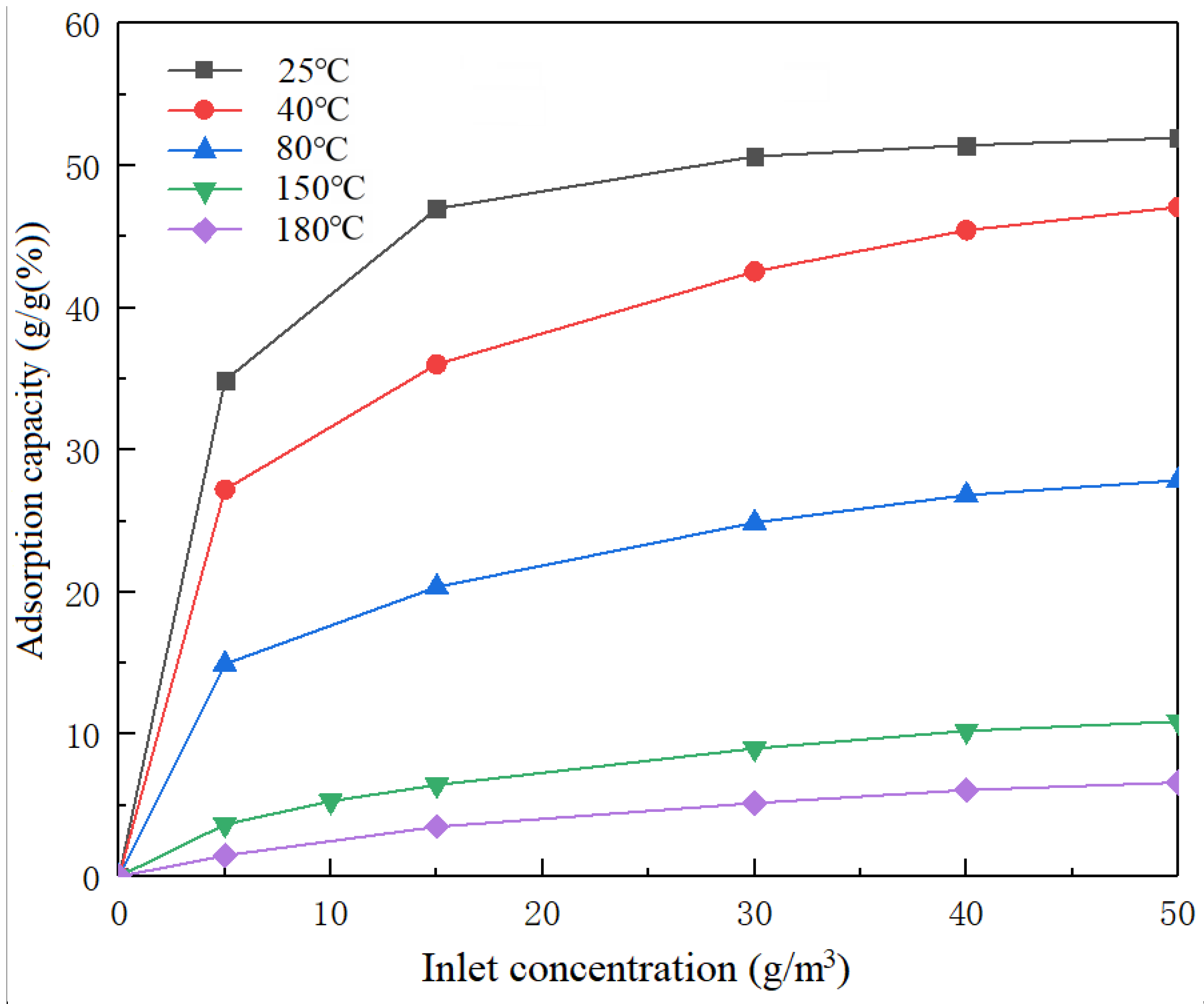
Figure 3 illustrates the toluene adsorption isotherm curve for a range of experimental temperatures.
A type I adsorption isotherm of monolayer adsorption was suitable for the adsorption isotherm curve [
20]. When the toluene concentration was 5 g/m
3, the equilibrium adsorption capacity of CTC 90 for toluene at 25 °C was 0.349 g/g. The equilibrium adsorption capacities were 0.2720, 0.1490, 0.03650 and 0.01470 g/g at experimental temperatures of 40, 80, 150 and 180 °C, respectively. When the concentration of toluene was 50 g/m
3, the equilibrium adsorption capacity at 25 °C was 0.5190 g/g. The equilibrium adsorption capacities were 0.4720, 0.2790, 0.1090 and 0.0660 g/g with experimental temperatures of 40, 80, 150 and 180 °C, respectively. The equilibrium adsorption capacity rose at the same temperature as the toluene concentration. When the toluene concentration was constant, the equilibrium adsorption capacity decreased with rising temperature. This was an exothermic reaction.
The curves in
Figure 3 were well fit by the Freundlich and Langmuir adsorption isotherm equations. The adsorption isotherms of toluene at 25, 40, 80, 150 and 180 °C on CTC90 activated carbon were fitted.
Figure 4 displays the results and the fitting curves.
qs in the Langmuir equation is the saturated adsorption capacity of a single molecular layer and
qs in
Table 3 is close to the measured value of the adsorption isotherm experiment. B is the empirical adsorption equilibrium constant of the Langmuir equation. Its value indicates the adsorption capacity of gas molecules on the solid surface. The b value in
Table 3 decreased with the rise in temperature, which was consistent with the conclusion that the equilibrium adsorption capacity decreased as temperature rose in the experiment. In the Freundlich equation, the constant k is related to the characteristics and temperature of the adsorbent and the adsorbate, and the constant n is related to temperature. Both can characterize the adsorption capacity of an adsorbent to an adsorbate, and these two parameter values also decrease with an increase in temperature. The R
2 of the Freundlich isotherm equation of activated carbon adsorbing toluene was basically higher than the Langmuir isotherm equation, and it was more stable, basically above 0.99. Therefore, with activated carbon adsorbing toluene, the Freundlich isotherm equation performed better than the Langmuir isotherm equation.
3.2. Working Capacity Curve
The equilibrium adsorption capacity was not zero when the desorption temperature reached equilibrium, so the maximum working capacity was used as the evaluation factor in practical applications. The working capacity was defined as the difference between the equilibrium adsorption capacity of an adsorbate of a certain concentration at a specific adsorption temperature and the equilibrium adsorption capacity of the corresponding equilibrium concentration when gaseous pollutants condense at a specific desorption temperature. The intake air concentration was taken as the abscissa and the working capacity as the ordinate. The adsorption temperature of activated carbon is 25 °C, and the desorption temperature is 150 °C. The working capacity under these working conditions was obtained by subtracting the equilibrium adsorption capacity corresponding to the toluene condensation temperature under the desorption temperature from the equilibrium adsorption capacity under each intake concentration.
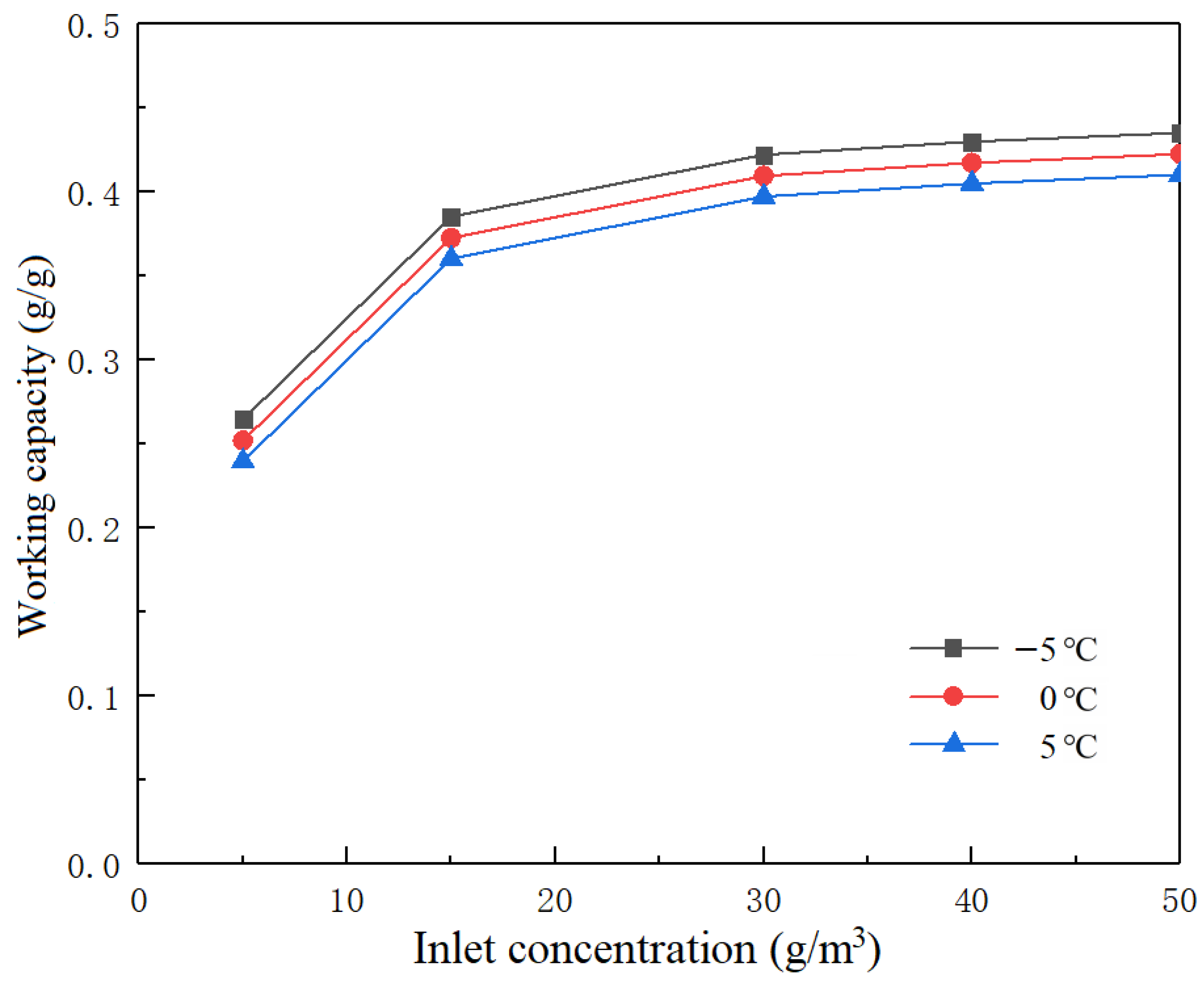
Figure 5 shows the maximum working capacity curve of activated carbon to toluene at different concentrations under different inlet air concentrations.
When the inlet toluene concentration of the adsorption treatment gas was 5 g/m3 and the condensation temperatures of the circulating gas were −5, 0 and 5 °C, the working capacities of toluene were 0.2650, 0.2520 and 0.2400 g/g, respectively. When the inlet toluene concentration was 15 g/m3 and the condensation temperatures were −5, 0 and 5 °C, the working capacities of toluene were 0.3850, 0.3730 and 0.3600 g/g, respectively. When the inlet toluene concentration was 30 g/m3 and the condensation temperatures were −5, 0 and 5 °C, the working capacities of toluene were 0.4220, 0.4100 and 0.3970 g/g, respectively. When the inlet toluene concentration was 50 g/m3 and the condensation temperatures were −5, 0 and 5 °C, the working capacities of toluene were 0.4350, 0.4220 and 0.4100 g/g, respectively. When the concentration at the inlet of the treatment gas was between 5 and 15 g/m3, the maximum working capacity rose rapidly. The maximum working capacity of CTC-90 activated carbon tended to be consistent at toluene concentrations higher than 15 g/m3, particularly at concentrations higher than 30 g/m3.
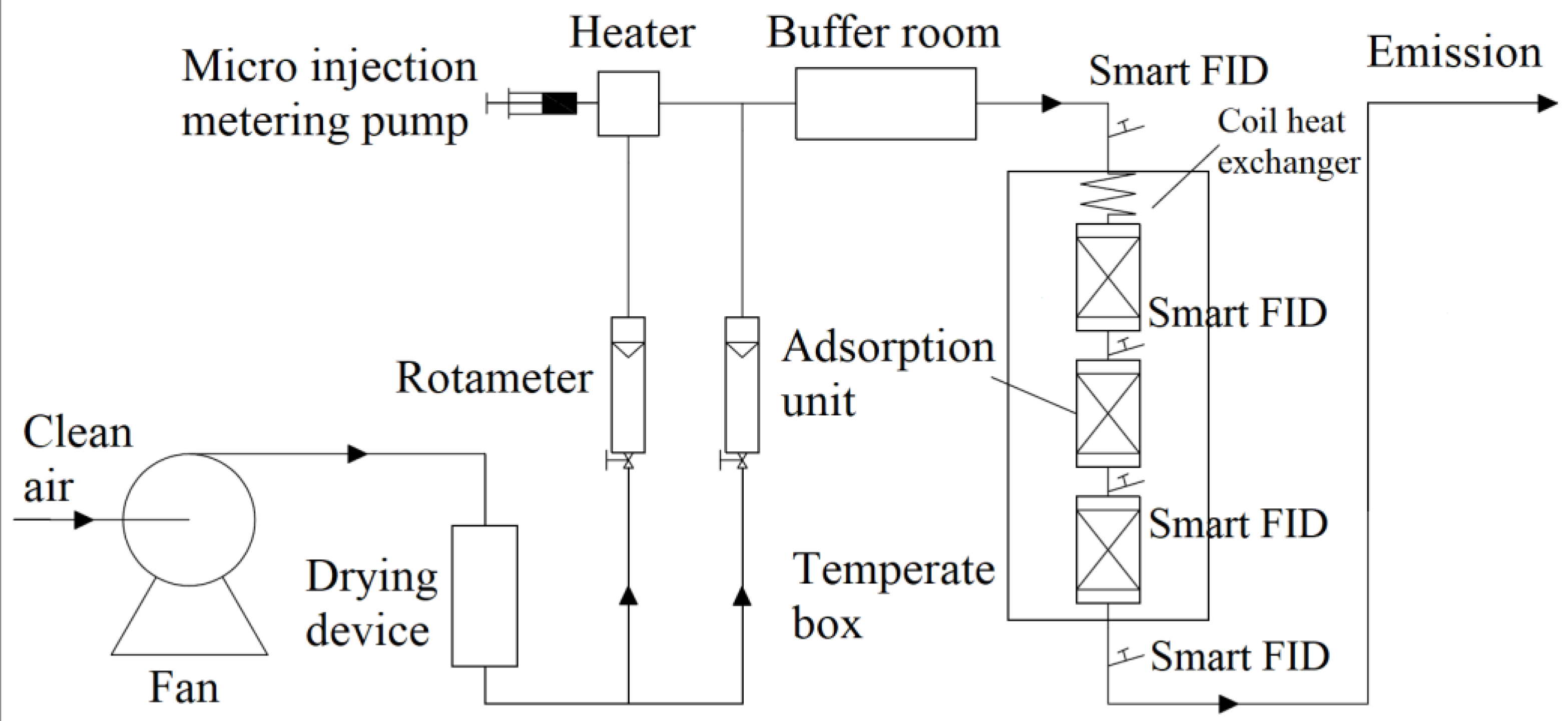
3.3. Analysis of Desorption Concentration and Desorption Amount of Closed Cycle Temperature Swing Desorption
In order to explore the impact of various inlet concentrations of toluene gas on the mass transfer process desorption by activated carbon in closed cycle temperature swing adsorption, we designed four different working conditions. The inlet concentrations were 0, 2, 5 and 10 g/m
3. We used an FID to monitor the toluene gas concentration at the outlet of each bed. The desorption unit was composed of three layers of desorption columns.
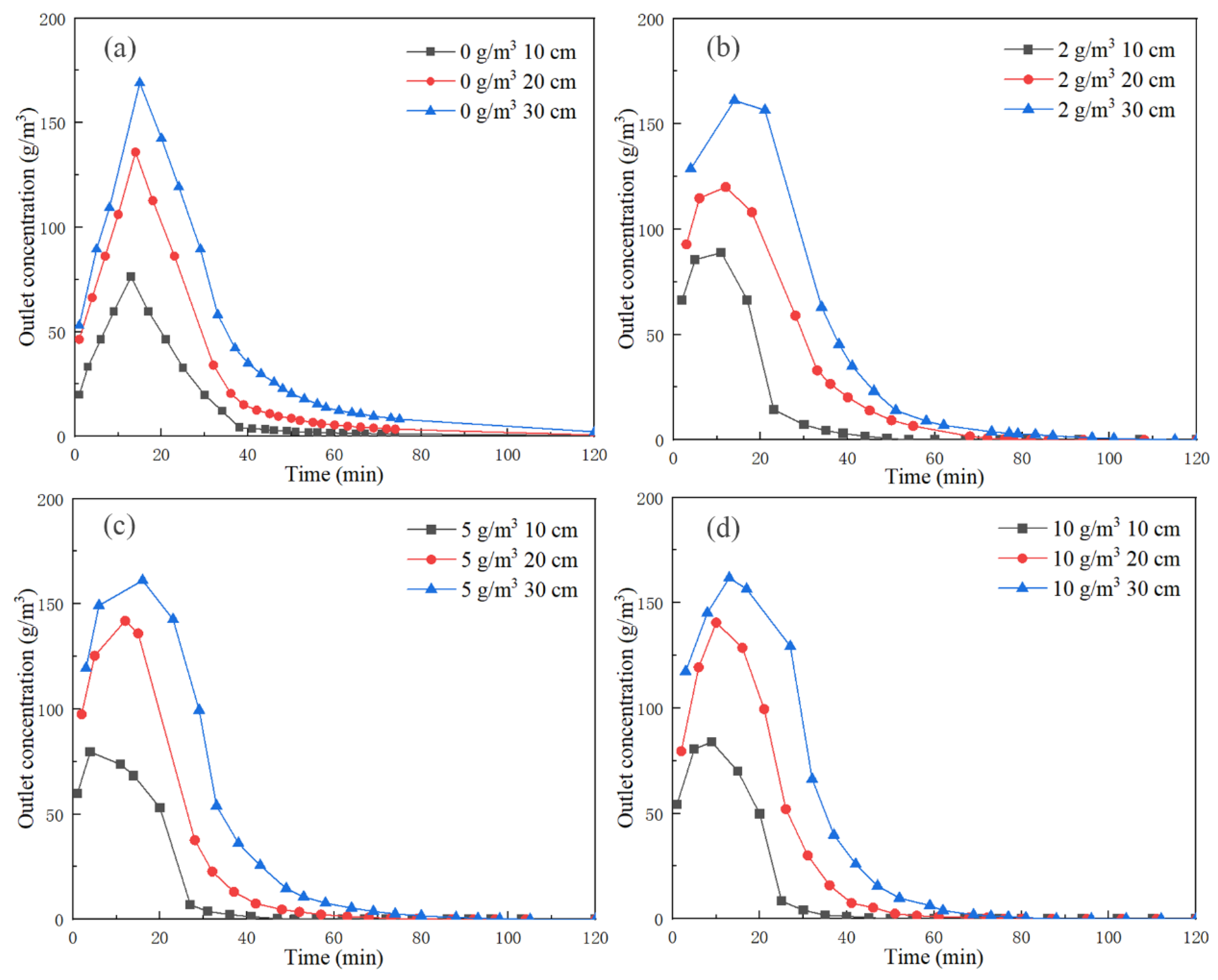
Figure 6 shows the outlet concentration curve at the outlet of each bed with various background concentrations (background concentration removed).
In the desorption concentration curves of all background concentrations, the toluene concentration at the outlet section of each bed rapidly rose to a peak, then dropped rapidly and finally stabilized at the background concentration. The toluene outlet concentration measured at 10 cm reached the peak in the shortest time and had the lowest peak concentration. The outlet concentration at 10 cm was the lowest throughout this experiment, and the time to complete desorption and reach equilibrium was the shortest. The time to reach the peak concentration at 30 cm was the longest and the peak concentration was the highest. The outlet concentration at 30 cm was always the highest throughout the experiment, and the time to complete the desorption was the longest. This was due to the fact that during desorption of activated carbon, the rear end activated carbon bed continuously adsorbed a high concentration of toluene desorbed from the front end. After the desorption concentration of the active carbon bed at the front end dropped rapidly, the activated carbon bed at the rear end was no longer affected by the front end desorption, and completely began to desorb.
We compare the outlet concentration curves under different background concentrations in
Figure 6. With different background concentrations, the time to reach the peak value did not change significantly and the peak value did not increase or decrease significantly. With the continuous increase in background concentration, the time from complete desorption to the stabilization of equilibrium was shortened. This is because introducing toluene at a specific concentration for desorption increased the outlet concentration of the desorption equilibrium.
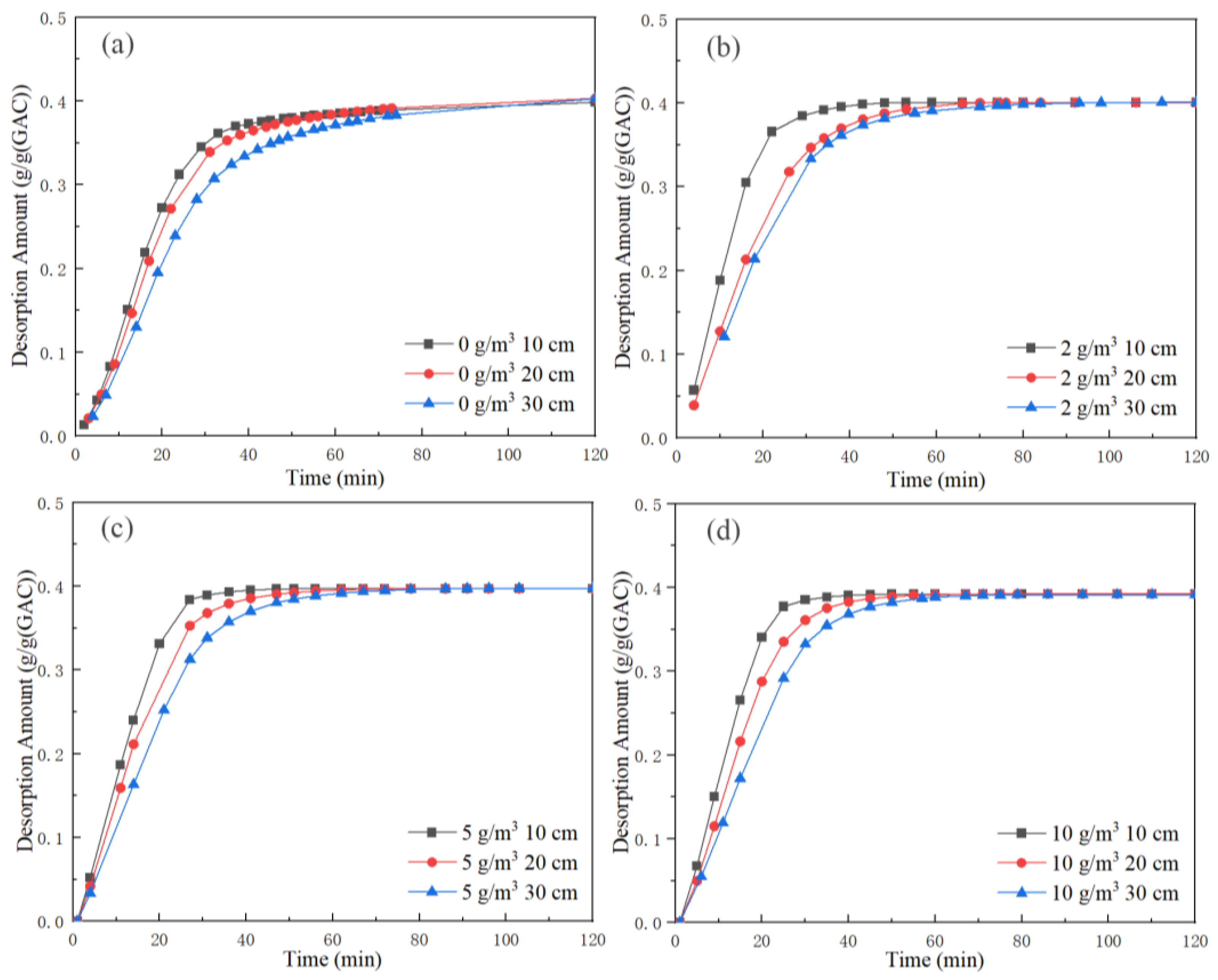
Through integrating the desorption concentration curve in
Figure 6, the toluene desorption amount at the outlet of unit activated carbon in each bed under different background concentrations was obtained, and the results are shown in
Figure 7. The toluene desorption amount per unit activated carbon of each bed started from zero, rose rapidly in a short time and then tended to be flat and stable. The toluene desorption amount per unit activated carbon at 10 cm was the highest, and the time to reach the inflection point and stabilize was the fastest. The toluene desorption amount per unit activated carbon at 30 cm was the lowest, and the time to reach the inflection point and stabilize was the slowest. Finally, the toluene desorption amount per unit activated carbon in each layer was equal.
Table 4 shows a summary of the results of toluene desorption per unit activated carbon at 120 min for each bed under different working conditions.
According to the analysis in
Table 4 and
Figure 8, under the same desorption temperature and the cross-section wind speed, with the increase background concentration, the time taken from the rapid rise to the inflection point was shorter, and the toluene desorption amount of unit activated carbon outlet gas in each bed layer was lower, but the change was very small.
3.4. Analysis of Average Rate and Instantaneous Rate of Closed Cycle Temperature Swing Desorption
The average desorption rate of unit activated carbon at 10 cm in 10 min is
(g/ (g • min)), at 10 cm in 20 min is
(g/ (g•min)), at 10 cm in 40 min is
(g/ (g•min)) and at 10 cm in 60 min is
(g/ (g•min)).
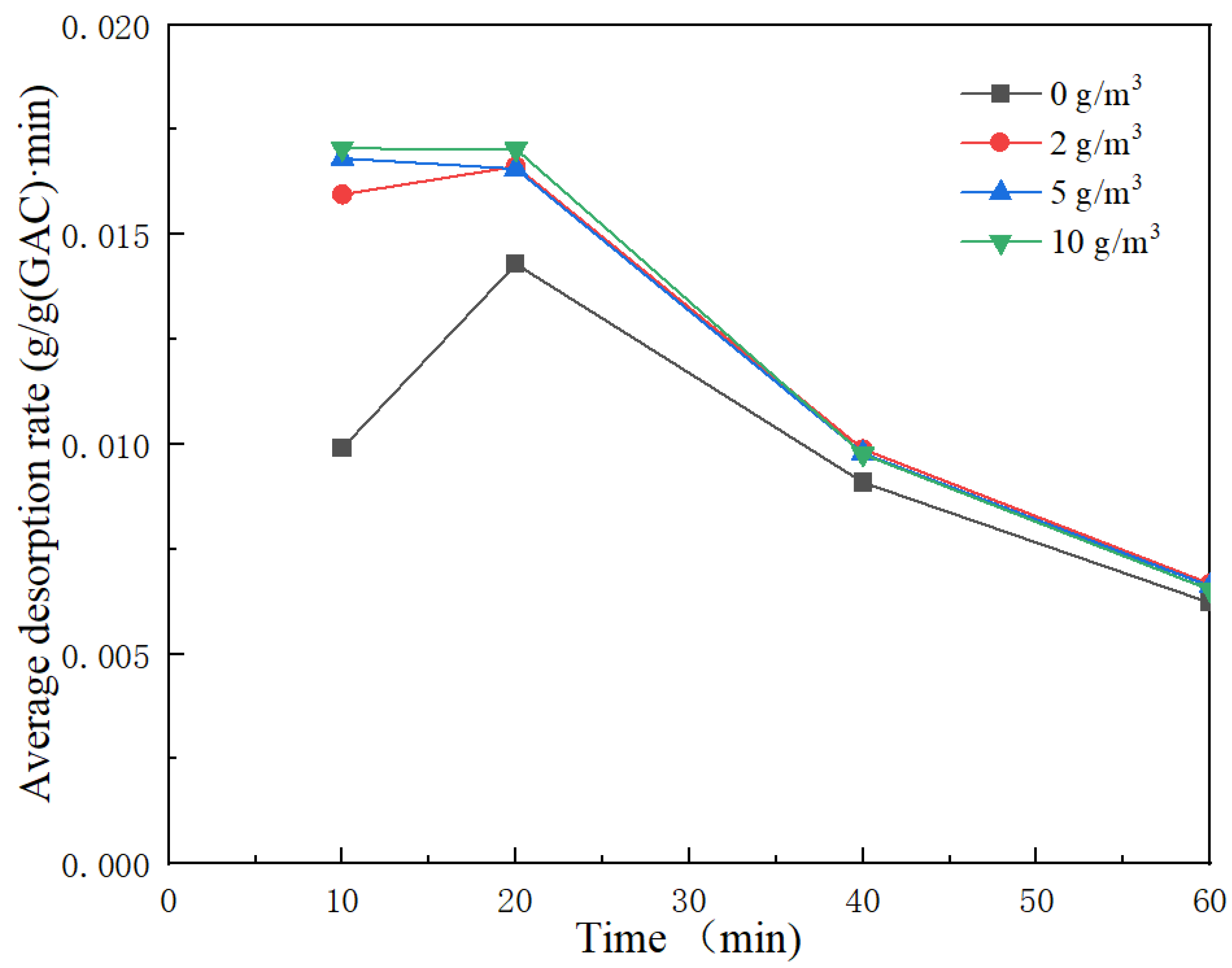
Figure 8 shows the average toluene desorption rate of the outlet gas at 10 cm at different times.
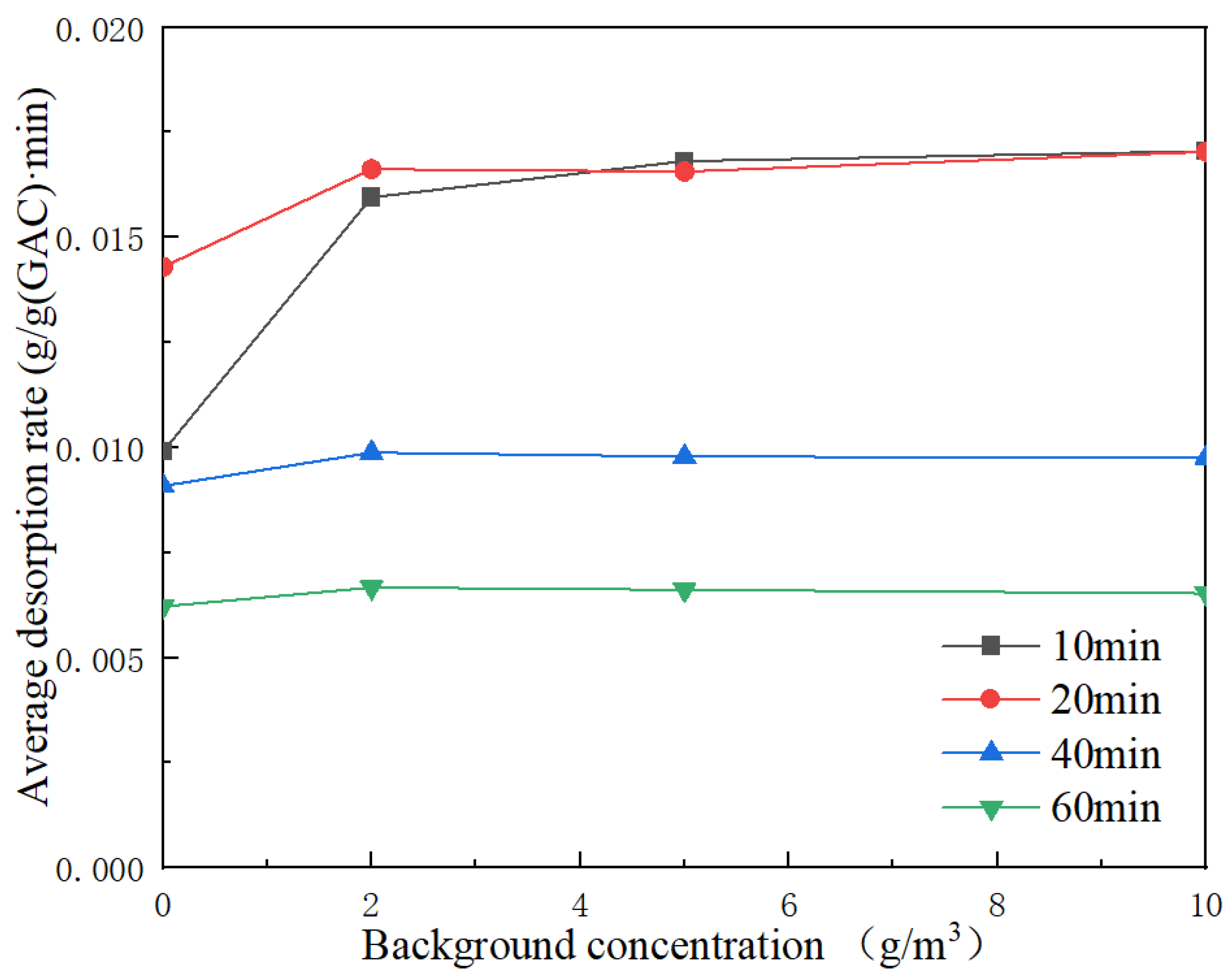
Figure 9 shows the average toluene desorption rate of outlet gas at 10 cm with different background concentrations.
Under the same working conditions, the at background concentrations of 0, 2, 5 and 10 g/m3 were 0.0099, 0.0160, 0.0168 and 0.0170 g/ (g•min), respectively. The at background concentrations of 0, 2, 5 and 10 g/m3 were 0.0143, 0.0166, 0.0166 and 0.0170 g/ (g•min), respectively. It can be concluded that at the same desorption temperature and the cross-section wind speed, and increase with the increase in background concentration. The 40 min average desorption rates, , at background concentrations of 0, 2, 5 and 10 g/m3 were 0.0091, 0.0099, 0.0098 and 0.0098 g/ (g•min), respectively. The 60 min average desorption rates, , at background concentrations of 0, 2, 5 and 10 g/m3 were 0.0062, 0.0067, 0.0066 and 0.0065 g/ (g•min), respectively. When the background concentrations were 0–2 g/m3, and also increased as the background concentration increased. and also decreased with the increase in background concentration and tended to be stable when the background concentrations were 2–10 g/m3.
When the background concentrations were 0 and 2 g/m
3, the average desorption rate at 10 cm first increased and then decreased, and the average desorption rate at 20 min was the highest. However, when the background concentrations were 5 and 10 g/m
3, the average desorption rate at 10 cm declined, and the average desorption rate at 10 min is the highest. Combined with the analysis of the adsorption isotherm in
Section 3.1, the adsorption isotherm barely changed when the concentration was between 2 and 10 g/m
3. Therefore, when the background concentrations were between 2 and 10 g/m
3, the average desorption rate in each period was almost the same.
The average desorption rate at 10 cm under different background concentrations showed a trend of first rising and then declining, and the maximum average desorption rate was . reflects the average desorption rate of the initial driving force at the beginning of desorption. reflects the average desorption rate when the initial driving force of desorption is large. reflects the average desorption rate of the driving force in the initial stage of desorption when the concentration of each bed gradually starts to decline gently.
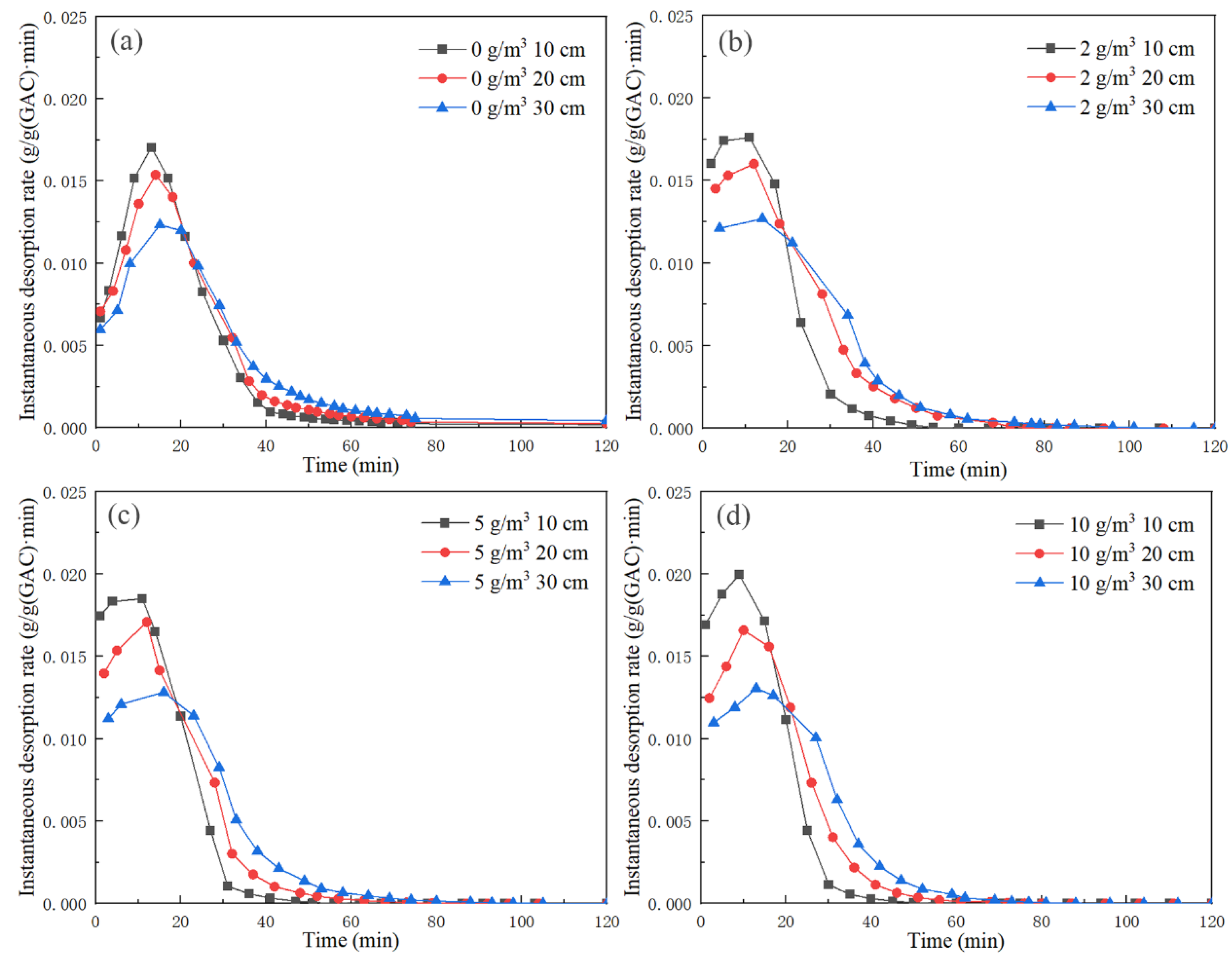
Through the differential of
Figure 7, the instantaneous desorption rate of unit activated carbon outlet gas in each bed under different background concentrations was obtained, and the results are shown in
Figure 10. At the beginning of the experiment, the instantaneous desorption rate of the unit activated carbon outlet gas in each bed was not zero, and it rapidly rose to the maximum
g/(g•min) in a short time, then rapidly dropped to a certain value and slowly became zero. The instantaneous desorption rate of unit activated carbon at 10 cm took the shortest time to reach
, and it was the highest from the beginning of the experiment to reaching
, and then dropped rapidly to the minimum until zero. The instantaneous desorption rate of unit activated carbon at 30 cm took the longest time to reach
, and it was the smallest from the beginning of the experiment to reaching
.
Table 5 shows
of each bed under different working conditions. At the same desorption temperature and the cross-section wind speed, the higher the background concentration, the larger the desorption
at each bed outlet and the faster the time to reach
. However, when the background concentration was too high, the desorption rate of activated carbon was reduced and the emission of toluene was too large.
3.5. Dynamic Simulation Analysis of Closed Cycle Temperature Swing Desorption
To investigate the effect of closed-cycle temperature swing desorption with different background concentrations on the migration of the adsorbate in the activated carbon layer, the desorption kinetics of toluene between activated carbon beds during desorption were studied by studying the desorption rate.
There are three typical types of kinetic factors, namely the quasi-first-order kinetic equation, the quasi-second-order kinetic equation and the Bangham adsorption rate equation. The differential form of the quasi-first-order kinetic equation is shown in Equation (2) [
21,
22].
Through the integral of Equation (2), we obtain Equation (3):
The differential form of the quasi-second-order kinetic equation is shown in Equation (4) [
23].
Through the integral of Equation (4), we obtain Equation (5):
The differential form of the Bangham desorption rate equation is shown in Equation (6) [
21,
22].
Through the integral of Equation (6), we obtain Equation (7):
where
qt represents the desorption amount (g/g) at time
t,
qe represents the desorption amount at equilibrium (g/g),
ka represents the quasi-first-order desorption rate constant (min
−1),
kb represents the quasi-second-order desorption rate constant (min
−1),
kc represents the Bangham desorption rate constant (min
−n) and
n represents the Bangham constant.
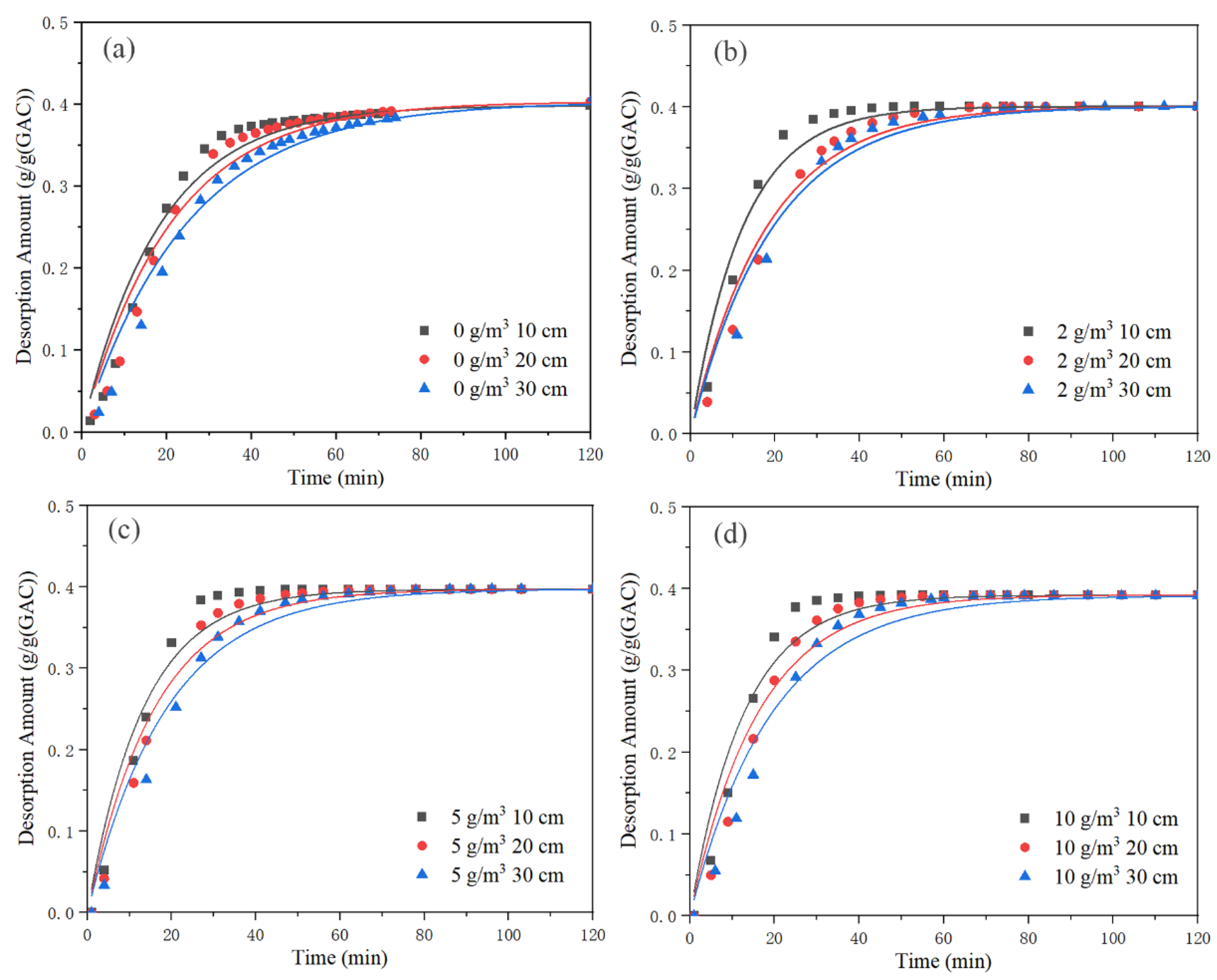
Figure 11 shows the fitting of quasi-first-order kinetic equation for each bed with different background concentrations. The fitting parameters for the quasi-first-order kinetic equation are displayed in
Table 6.
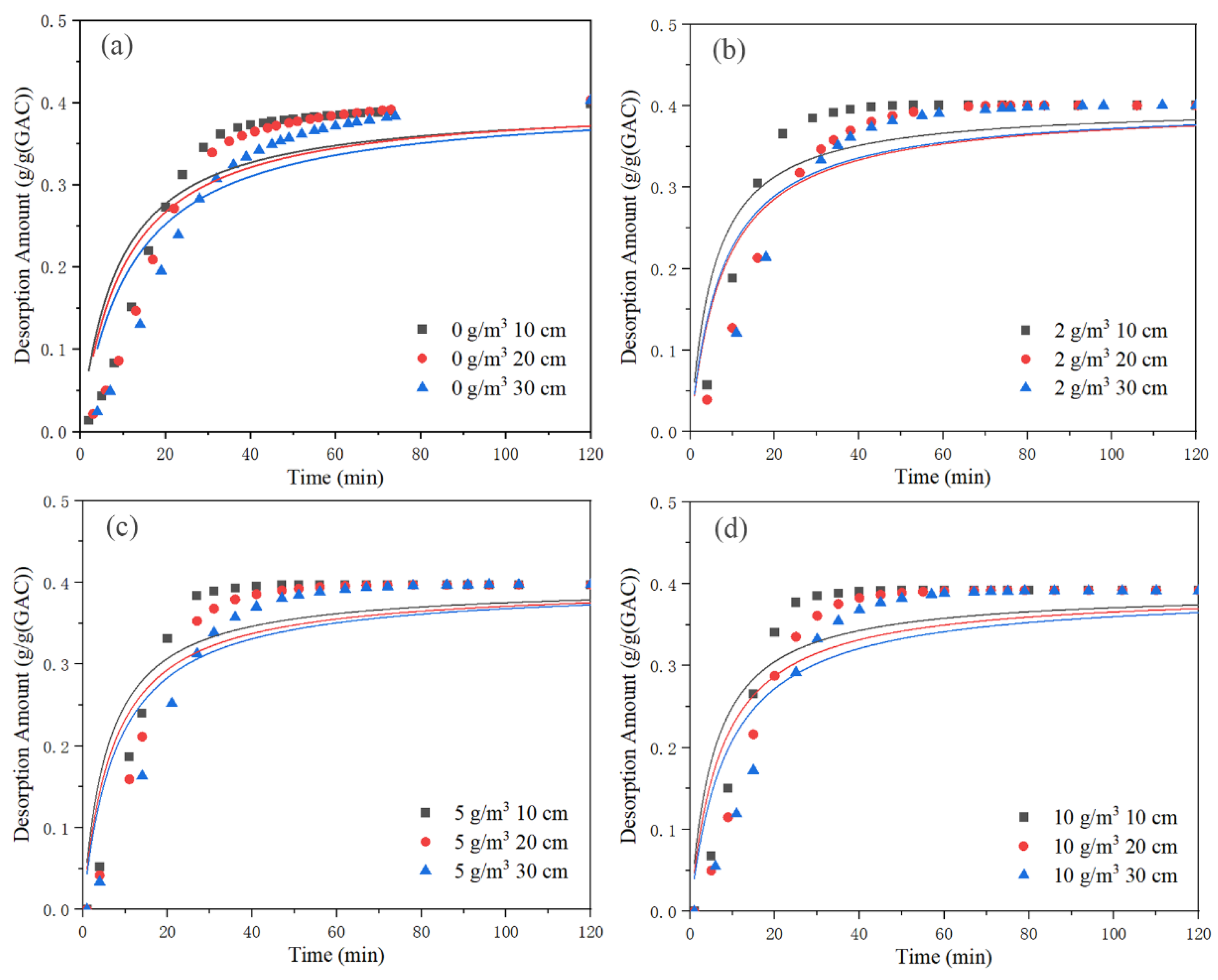
Figure 12 depicts the fitting of quasi-second-order kinetic equation. The fitting parameters for the quasi-second-order kinetic equation are displayed in
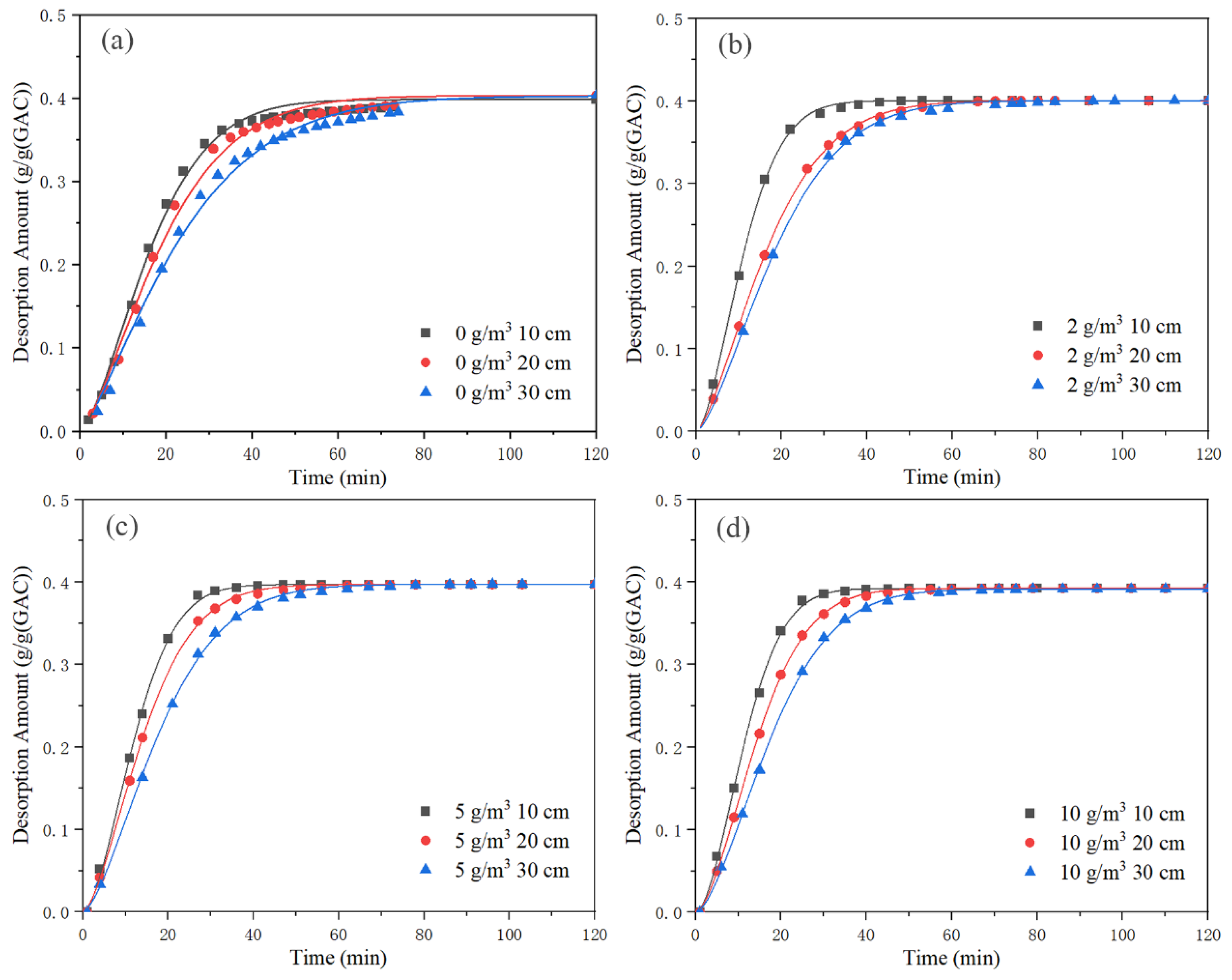
Table 7. The Bangham desorption rate equation’s fit is depicted in
Figure 13. The fitting parameters for the Bangham desorption rate equation are displayed in
Table 8. The value of
qe was taken from the data in
Table 4. According to
Figure 11 and
Table 6, the R
2 value of the quasi-first-order kinetic equation was greater than 0.9591, and the fit was good. According to
Figure 12 and
Table 7, the R
2 value of the quasi-second-order kinetic equation was less than 0.90, and the fit was poor. According to
Figure 13 and
Table 8, the R
2 value of the Bangham desorption rate equation was greater than 0.99, and the fit was the best out of the three equations. The three kinetic models’ desorption rate constants rose initially, then fell as background concentration rose. A background concentration of 2 g/m
3 resulted in the highest desorption rate constant. However, when the saturated activated carbon was desorbed by clean gas, the desorption rate constant was low. The Bangham constant increased with the increase in background concentration. The desorption rate constants of the three kinetic models decreased with the increase in activated carbon bed length. This is similar to the analysis of average desorption rate and instantaneous desorption rate. It shows that under the conditions of closed cycle adsorption, the concentration polarization layer of activated carbon was broken when a certain concentration of toluene was purged. While the external diffusion mass transfer effect increased, the internal mass transfer resistance decreased.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}