Abstract
Nuclear fusion is a phenomenon that is well known within the nuclear physics community as a viable option for alternative energy as many natural gases and fossil fuels are phased out of commercial use. Deuterium and tritium fusion reactions are currently the leading candidates for nuclear fusion, with a major limiting factor being a means to produce tritium on an industrial scale. Lithium-6 is a well-known isotope that can produce tritium and helium following a fission reaction with a neutron. Unfortunately, the lithium-6 enrichment methods are limited to the COLEX process, which leaves behind an alarming amount of mercury waste as a potential environmental contaminant. Deep eutectic solvents are believed to be a potential alternative to lithium isotope separations due to the ease of generation, in addition to the minimum environmental waste generated when these solvents are employed. Previous studies have suggested that deep eutectic solvents are capable of separating lithium isotopes by utilizing a 2-thenoyltrifluoroacetone and trioctylphosphine oxide system that can biphasically react with a buffered solution containing lithium chloride. This system displays a separation factor of 1.068, which when compared to the 1.054 separation within the COLEX process, makes it a potential candidate for lithium-6/7 separation. Within this study, we investigate this system in comparison to two newly synthesized deep eutectic solvents and find that within these acetylacetone-based systems, little isotopic separation is observed. We investigate these systems both experimentally and computationally, showing the different lithium cation affinities, in addition to proposing how the electron-donating or -withdrawing nature can influence these systems.
1. Introduction
Lithium is an alkali metal that has two stable isotopes, lithium-7 (92.4%) and lithium-6 (7.6%), within the Earth’s crust [1]. With many applications across disciplines, lithium is most notably known for lithium-ion batteries [2]. Lithium is also present in other industries related to ceramics and glasses, greases and lubricants, polymers, and pharmaceutical agents [3]. One subset of lithium utilization is within nuclear fusion as a means of tritium production [4]. If bombarded by neutrons, lithium-6 decomposes into both helium and tritium, which, with deuterium, is vital for the future of nuclear fusion [5]. Of equal necessity, lithium-7 also plays a role within nuclear fission where it acts as a cooling agent in the form of lithium hydroxide [6]. In this regard, 99.95% lithium-7 hydroxide is present in low concentrations to counteract the corrosion from boric acid, which is generated from the boron-10 fusing with a neutron and then subsequently reacting with water [7]. These applications within nuclear chemistry necessitate a means to isolate lithium-6 and lithium-7 as the natural isotopic ratio inhibits any nuclear reaction occurring.
Dating back to the early 1950s, the COLEX process has been the mainstay within industrial-scale lithium isotope separations [8]. The major limiting factor for this process is the mercury biproduct produced by the flux between mercury and aqueous lithium hydroxide leading to the separation. Other examples within the literature on laboratory-scale lithium isotope extractions, with meaningful separation factors above that of 1.03, can be mediated by the crown ethers within organic media to facilitate an extraction by a biphasic reaction with an aqueous media containing a buffered solution of lithium [9,10]. Despite having a remarkable separation factor of 1.046, which is comparable to the value of that shown within the COLEX process (1.054), there are limitations to this means of separation associated with the expense and sensitivity of the crown ethers. Attempts have been made to utilize electrochemical dialysis and electromigration; however, both methods are limited by either using molten lithium, which is highly corrosive, or the use of aqueous or organic solvent coordinating to lithium too strongly, preventing migration from taking place [11,12].
A proposed alternative to the previously mentioned means of separation is the use of deep eutectic solvents (DESs) [13]. DESs are a subset of ionic liquids known for their green and sustainable characteristics [14]. Although there are four different subsets within DESs, many of the application-based developments utilize a hydrogen bond donor and a halide salt due to the ease of formation and stability of the DES. For this reason, deep eutectic solvents are believed to offer a solution to this problem. Their aqueous stability, in addition to their organic nature, allows for biphasic reactivity, which can occur at a large industrial scale, where a metathesis reaction could take place between a cation from the DES and a cation within the aqueous media. Furthermore, this reactivity could be optimized by utilizing acid–base chemistry for the binding or extraction of a given isotope from the DES.
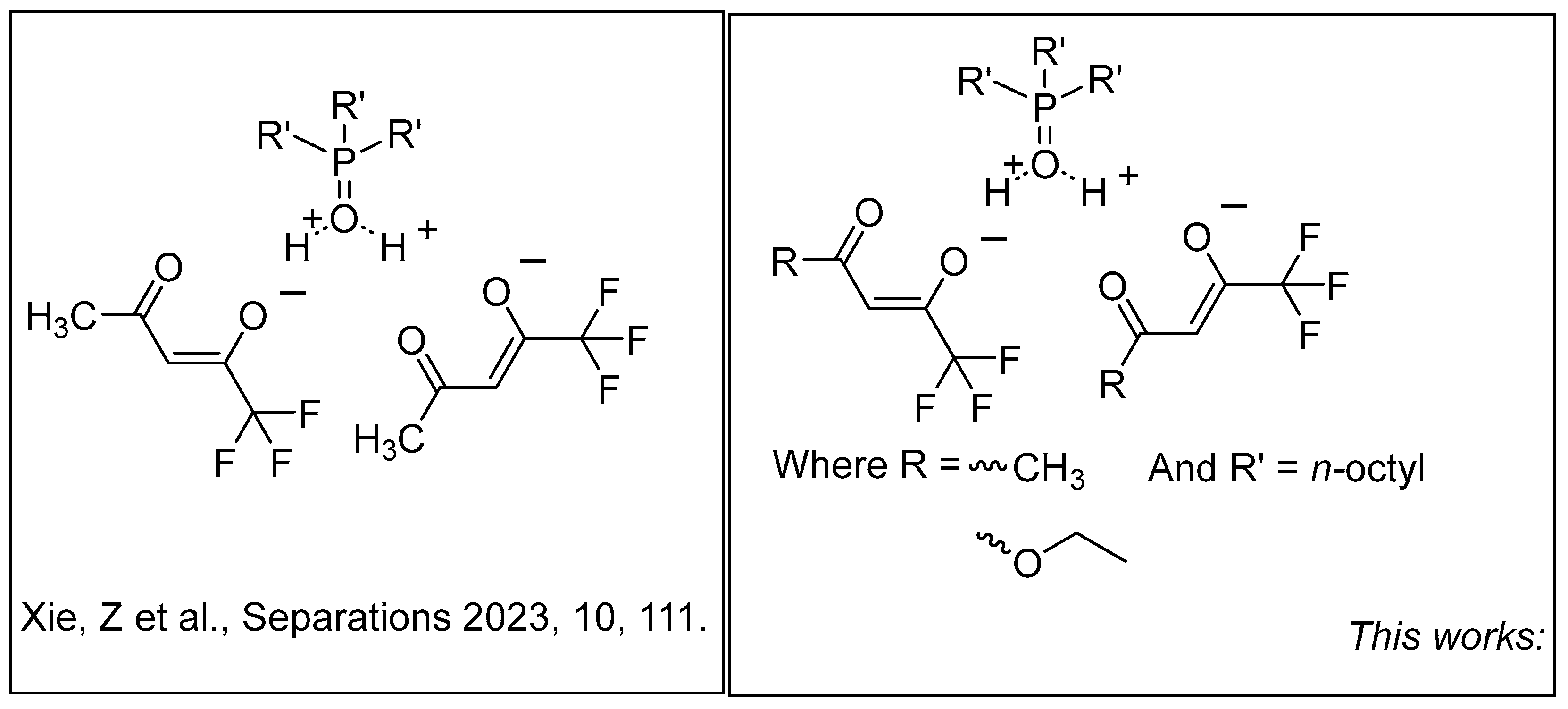
DESs have primarily previously been shown to be capable of extracting and recycling lithium present in batteries [15]. Recently, Jiang has shown that DESs may potentially be used to extract lithium-6 versus lithium-7 by utilizing a 2-thiophenacetylacetone and trioctylphosphine oxide-based DES (Figure 1) [16]. These monomers exhibit properties similar to crown ethers in that there is a specific binding constant associated with the β-diketone-based ligand. What separates DESs from crown ethers is their innate stability and ability to be recycled and regenerated up to six extractions, as shown by Jiang. Within this study, we will investigate two newly synthesized DESs based upon the previously studied system that bound lithium. We will interrogate both experimental and computational means to gain an understanding of lithium isotope separation within these DES-based systems.
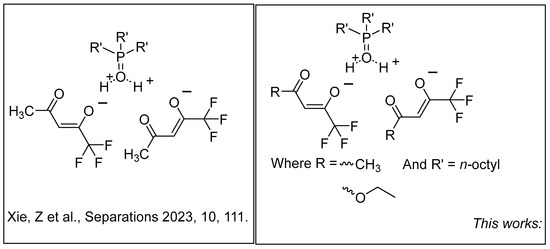
Figure 1.
Structural depiction of previously published HTTA/TOPO-based systems and newly generated DES systems featuring varying R groups [16].
2. Materials and Methods
2.1. Reagents and Chemicals
The 2-thenoyltrifluoroacetone (99%), 1,1,1-trifluoro-2,4-pentanedione (98%), ethyl 4,4,4-trifluoroacetoacetate (99%), tri-octyl-phosphine oxide (TOPO, 99%), lithium chloride (99%), and ammonium chloride (99%) were all ordered from Sigma Aldrich (Rayleigh, NC, USA). The lithium chloride mass spectrometry isotope standard was ordered from VWR (Charlotte, NC, USA). The distilled water was purified with a Sartorius Arium Pro (Bohemia, NY, USA) (0.055 µS/cm).
2.2. Preparation and Characterization of DES
HTTA/TOPO was synthesized according to the literature procedure. To a flask containing TOPO (750 mg, 1.94 mmol), either 1,1,1-trifluoro-2,4-pentanedione (567 µL, 3.88 mmol) or ethyl 4,4,4-trifluoroacetoacetate (567 µL, 3.88 mmol) was added. The flask was then heated to 40 °C for 3 h, generating the formation of 1,1,1-trifluoro-2,4-pentanedione/TOPO or ethyl 4,4,4-trifluoroacetoacetate/TOPO in a quantitative yield verified by NMR spectroscopy. The NMR spectroscopy was run on a Magritek Spinsolve 60 spectrometer (Malvern, PA, USA), and the IR was run on a JASCO FTIR 6100 (Atlanta, GA, USA). The ICP-MS was run on a Thermofisher Element 2 HR-ICP-MS (Florence, SC, USA) instrument calibrated using a lithium chloride mass spectrometry isotope standard from VWR with a measured 7Li/6Li ratio of 12.17523 and a sensitivity down to 5 ppb.
2.3. Lithium Extraction Experiment
A solution of 25 ppm lithium chloride in an ammonium buffer was prepared according to the optimized literature procedure [16]. To this solution, 1,1,1-trifluoro-2,4-pentanedione/TOPO or ethyl 4,4,4-trifluoroacetoacetate/TOPO was added in a 4:1 buffer to the DES ratio. The now biphasic mixture was stirred for 450 rpm at 40 °C for 6 h. Following stirring, the solution was allowed to cool to room temperature and was centrifuged at 3000 rpm for 5 min. Following centrifugation, the aqueous layer was removed and sent for ICP-MS analysis. To the organic, assuming the same volumes, a 4:1 mixture of 0.1 M HCl was added, and the solution was mixed by inversion followed by sonication. Following the mixing, the solution was centrifuged down at 3000 rpm for 5 min and the aqueous layer used for extracting the organic layer was sent for analysis.
2.4. Computational Studies
The optimization and frequency calculations were run with the functional B3LYP and basis sets 6–311 (g). The lithium cation affinities were calculated using the optimized structures energy to measure the enthalpy of formation upon treating each DES with one equivalent of lithium cation in the gas phase. The depictions of each structural optimization were visualized within the VESTA package 3.5.8.
3. Results
To begin this investigation, we first synthesized two new DESs using TOPO in a 1:2 ratio with 1,1,1-trifluoro-2,4-pentanedione and ethyl 4,4,4-trifluoroacetoacetate to form 1,1,1-trifluoro-2,4-pentanedione/TOPO and/or ethyl 4,4,4-trifluoroacetoacetate/TOPO DES. Our strategy for investigating these molecules was to alter the donicity of the functionalities surrounding the β-diketone-based molecule to gain a further understanding of the effects that the electron-withdrawing or -donating groups had on the separation performance when compared to previously published structures, with the goal of moving away from the potentially toxic, fluorination-based DES. It is well known that acetylacetone ligands will form enolates within solvents, making them more protic and more prone to form the acetylacetonate-based anion. However, the acetylacetone-based ligand with additional electron donicity from the R substituents is believed to form this enolate less readily in comparison to an acetylacetone-based ligand with more electron withdrawing groups upon it.
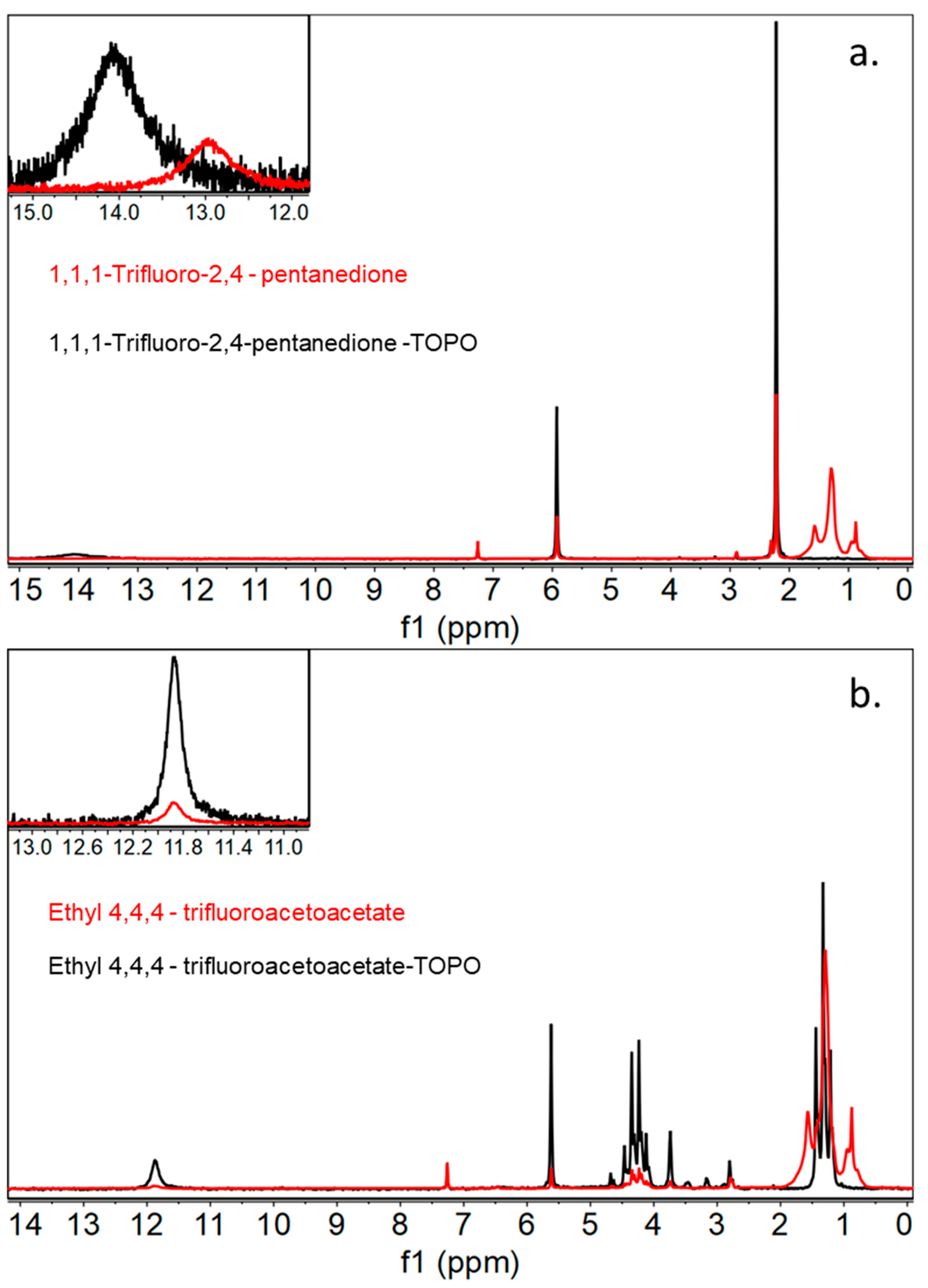
Characterization of these newly generated DESs by 1H NMR showed that there is indeed an acidic proton associated with the β-diketone-based ligand that is present at approximately 12 ppm for both 1,1,1-trifluoro-2,4-pentanedione/TOPO and ethyl 4,4,4-trifluoroacetoacetate/TOPO (Figure 2). In addition, IR spectroscopy displayed a nearly identical stretch frequency of 3017 cm−1 for 1,1,1-trifluoro-2,4-pentanedione/TOPO and 3018 cm−1 for or ethyl 4,4,4-trifluoroacetoacetate/TOPO for the -OH group attached to the acetylacetonate ligand, indicating relatively equal acidities between the two protons. Attempts were made to crystallize the monomer within the deep eutectic solvents by cooling the solution to −20 °C, but these attempts failed due to the high degree of freedom within the DES, leading to difficulties generating a single crystal. Upon treatment with a buffered solution containing lithium chloride and then centrifuging the deep eutectic solvent, removing the aqueous layer, it was shown that the specific proton had indeed vacated the DES, leading us to believe that lithium binding did in fact occur.
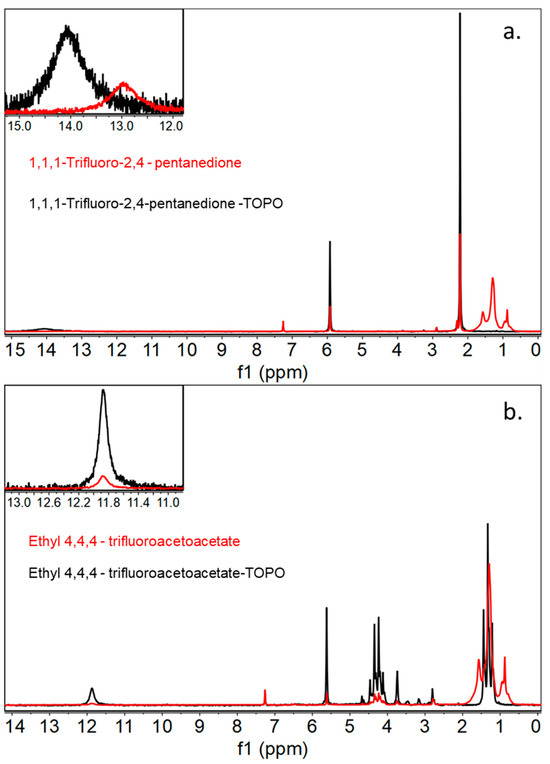
Figure 2.
1H NMR spectra of monomer ethyl 4,4,4-trifluoroacetoacetate in red compared to the generated DES ethyl 4,4,4-trifluoroacetoacetate/TOPO spectra in black, with an inset highlighting the generation of an acidic proton peak (a) and 1H NMR spectra of monomer 1,1,1-trifluoro-2,4-pentanedione in red compared to the generated DES 1,1,1-trifluoro-2,4-pentanedione/TOPO in black (b), with an inset highlighting the generation of an acidic proton peak.
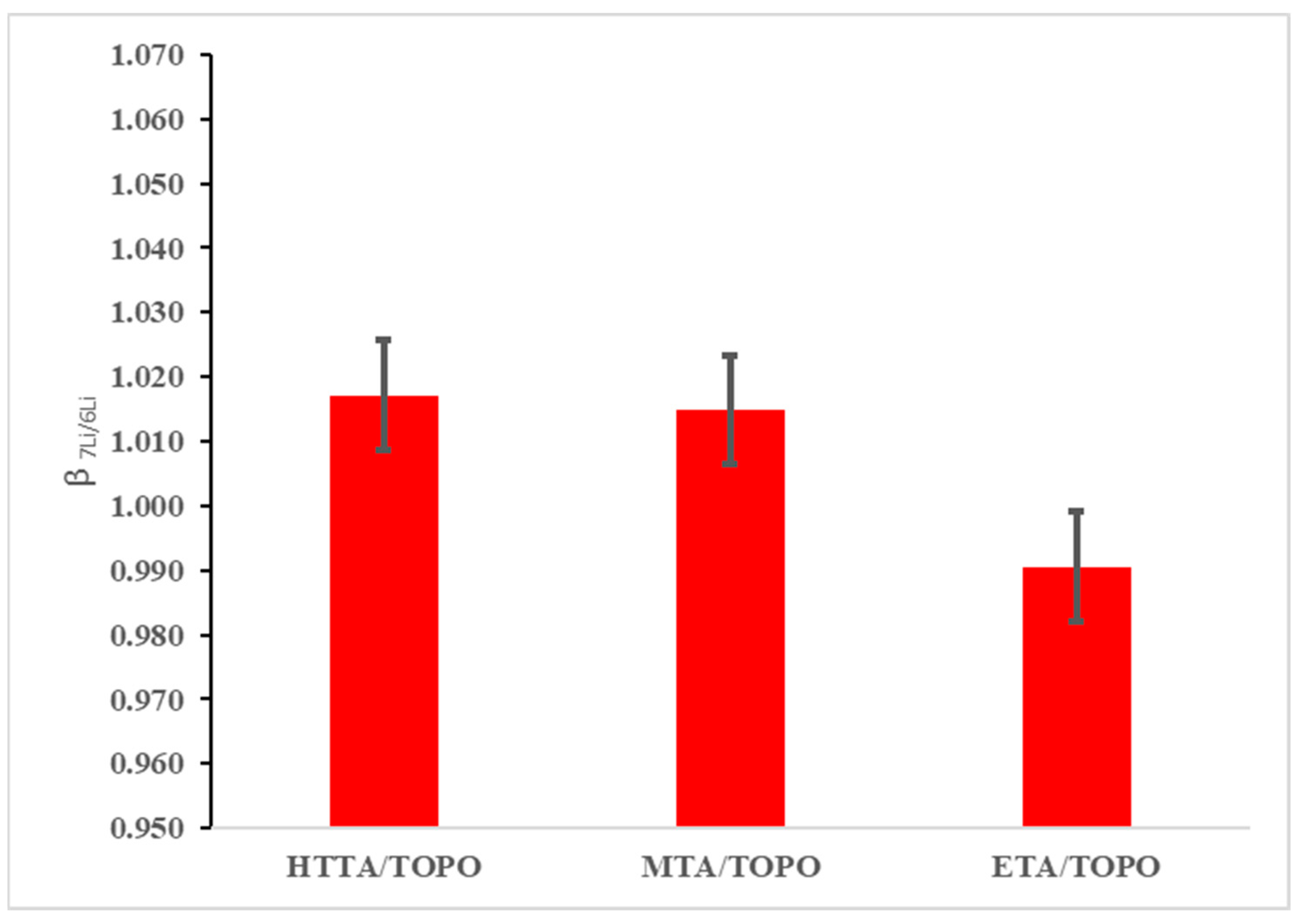
To compare the ability of the newly synthesized 1,1,1-trifluoro-2,4-pentanedione and ethyl 4,4,4-trifluoroacetoacetate/TOPO to bind lithium-6 versus lithium-7 to a previously published DES, a series of separation experiments were completed. Each experiment evaluated the difference in separation factors as a means to compare the enrichment efficiencies by a ratio of the natural abundance of an isotope compared to the new ratio following extractions for each isotope. The isotope separation experiments were run at 40 °C for 6 h following the previous literature procedures, which optimized the experimental conditions [16]. Following extraction, ICP-MS analysis was run for each isotope separation, comparing 1,1,1-trifluoro-2,4-pentanedione and ethyl 4,4,4-trifluoroacetoacetate/TOPO to TTA/TOPO, which was previously reported to have a separation factor of 1.068 (Figure 3). After three separate reactions for each DES solvent, an average of the separation factors was taken for each solvent, with the values calculated as 1.017, 1.015, and 0.991 for TTA/TOPO, 1,1,1-trifluoro-2,4-pentanedione, and ethyl 4,4,4-trifluoroacetoacetate/TOPO, respectively, all of which are significantly lower than the values, with each result conflicting with those previously reported in the literature.
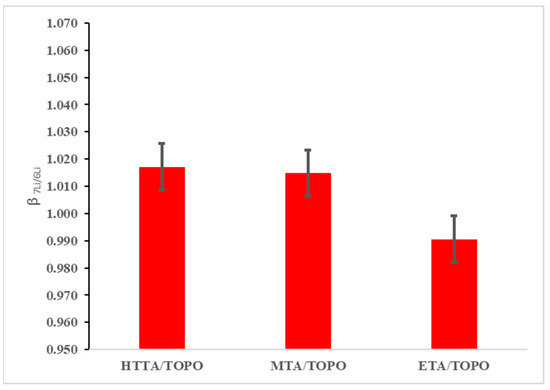
Figure 3.
Results from the 1,1,1-trifluoro-2,4-pentanedione and ethyl 4,4,4-trifluoroacetoacetate/TOPO system compared to previous methods of lithium isotope separation.
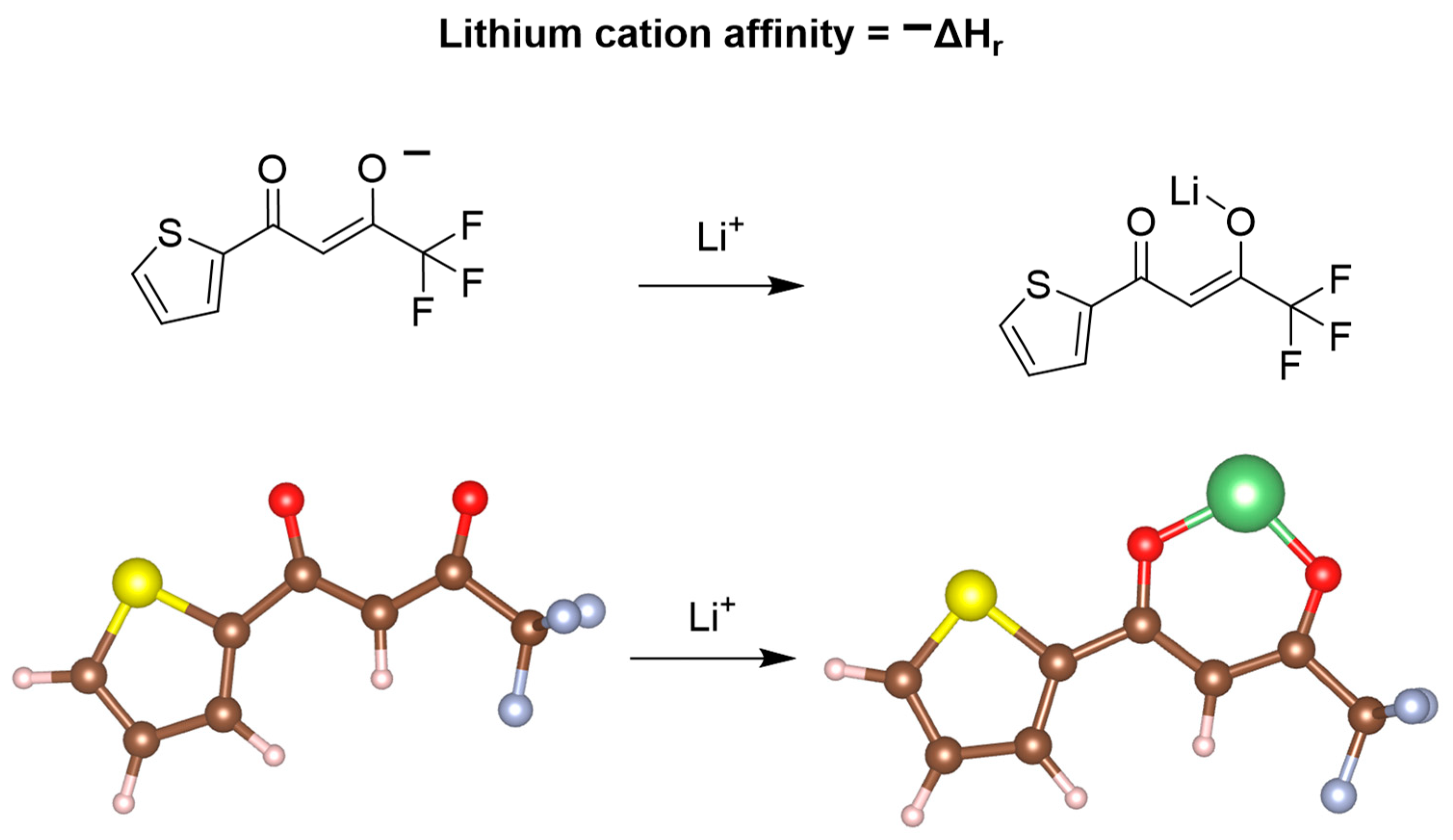
To explore the mechanism of these extractions and how we could potentially improve upon these results, we investigated this system utilizing lithium-ion affinity calculations (Figure 4). Due to TOPO regularly decoordinating experimentally and each acetylacetonate-based ligand favoring the formation of dimeric-based structures with lithium in the gas-phase calculations, optimizations were completed for TTA, 1,1,1-trifluoro-2,4-pentanedione, and ethyl 4,4,4-trifluoroacetoacetate, all in the absence of TOPO. Upon computing the relaxed gas-phase structures, lithium cation affinity calculations were completed for each system. When comparing the three lithium-ion affinities in the gas phase, we found that 1,1,1-trifluoro-2,4-pentanedione had the highest lithium-ion affinity of −188.1 kcal/mol, in comparison to ethyl 4,4,4-trifluoroacetoacetate and TTA/TOPO having values of 180.6 and 180.1 kcal/mol, respectively. Intriguingly, these affinities did not reflect the electron donicity of the flanking R group surrounding the acetylacetonate-based ligand, indicating that the induction effects may not be sufficient for predicting the experimental outcomes. Despite this, these lithium cation affinities do support a thermodynamically favored process allowing for lithium extraction from aqueous media. When changing the calculation to account for lithium-6 versus lithium-7, we found that there was no difference in the lithium cation affinity for all three DESs, eliminating the idea of a potentially thermodynamically favored isotope. These findings may be reflective of lithium-6/7 possessing relatively identical binding through what is believed to be a mostly electrostatically driven mechanism.
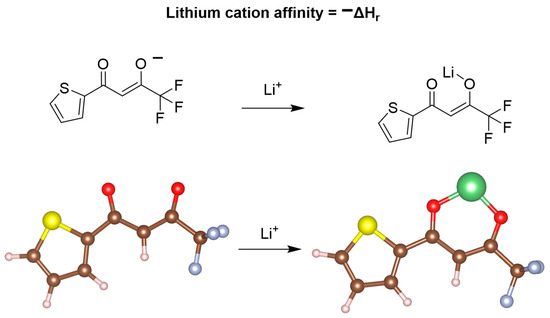
Figure 4.
Schematic representing the lithium cation affinity calculations and the associated structures following the geometric optimization calculations, as visualized through VESTA.
4. Discussion
Deep eutectic solvents, as a means of isotope separation, show promise when in comparison to the use of expensive crown ether reagents or the toxic conditions utilized in the COLEX process. With high extraction efficiencies in terms of lithium from aqueous conditions, the idea of tuning the DES system to have different affinities for Li-6/7 seems to be within the realm of possibility for future investigations. Acetylacetone-based systems, despite having high extraction efficiencies, may not be the answer to this conundrum. Despite having a large amount of DES-based systems published within the literature, there is room for a vast improvement within the understanding of different affinities for lithium-6/7. Most notably, utilizing systems with some form of chelation has proved to be the best option for performing these extractions, as aqueous media sequesters lithium cations with strongly favorable solvation spheres. In addition to this, different anions associated with the lithium may be worth investigating as various lithium salts display different behavior within aqueous media. Utilizing carbonates, sulfates, and other polyanionic species may be beneficial in terms of differentiating between the two isotopes as the coordination sphere may differ within these polyatomic anions. Additionally, the buffered solution may be an important factor as high levels of acidity favor lithium localizing aqueous media as the acidic proton may dominate within the DES media. Furthermore, basic conditions have yet to be investigated, which will change the coordination environment for the lithium cations as the basic media may slow the extraction of lithium from the aqueous media, potentially leading to more selectivity.
5. Conclusions
The results obtained in this study show that DESs can readily be made utilizing a β-diketone/TOPO-based system with different flanking R groups to facilitate different electronic environments, which may influence the binding affinity for different chemical species. In addition, these DESs may be used to separate lithium from aqueous media. This study also shows the need for further development within DES systems for potential selectivity for lithium-6/7, as shown by the experimental separation factors of 1.017 for the previously published HTTA/TOPO system, 1.015 for the 1,1,1-trifluoro-2,4-pentanedione system, and 0.991 for the ethyl 4,4,4-trifluoroacetoacetate system. These results reflect poorly in comparison to other separation methods shown within the literature, as separation factors greater than 1.03 are believed to be feasible candidates for an industrial setting and as a potential replacement for the COLEX process. Theoretical work calculating the lithium cation affinities showed no energetic preference for binding either isotope using any of these investigated systems. However, differences in the cation affinity were displayed, with 1,1,1-trifluoro-2,4-pentanedione showing a 5.0 kcal superior affinity relative to the other systems, indicating that tuning the lithium chelator within these deep eutectic solvents may enhance the extraction efficiencies from aqueous media.
Author Contributions
J.E.S. performed the hands-on experiments and theoretical work within this study. K.D.M., D.A.H. and B.L.G.-D. developed the conceptualization, methodology, and supervised this study. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funds from LDRD-00193-2023 of the Laboratory Directed Research and Development (LDRD) program within the Savannah River National Laboratory (SRNL). This document was prepared in conjunction with work accomplished by Battelle Savannah River Alliance, LLC (BSRA) under Contract No. 89303321CEM000080 with the U.S. Department of Energy.
Data Availability Statement
Data are contained within the article.
Acknowledgments
We would like to offer deep thanks to the LDRD program for funding this investigation. In addition, we would like to acknowledge Lindsey Roy, Stephen Vicchio, and Jonathon Baker for their insight into the theoretical work within this investigation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Penniston-Dorland, S.; Liu, X.-M.; Rudnick, R.L. Lithium Isotope Geochemistry. Rev. Mineral. Geochem. 2017, 82, 165–217. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Talens Peiró, L.; Méndez, G.V.; Ayres, R.U. Lithium: Sources, Production, Uses, and Recovery Outlook. JOM 2013, 65, 986–996. [Google Scholar] [CrossRef]
- Alvarez, L.W.; Cornog, R. Helium and Hydrogen of Mass 3. Phys. Rev. 1939, 56, 613. [Google Scholar] [CrossRef]
- Oliphant, M.L.E.; Kinsey, B.B.; Rutherford, E. The transmutation of lithium by protons and by ions of the heavy isotope of hydrogen. Proc. R. Soc. Lond. Ser. A Contain. Pap. A Math. Phys. Character 1933, 141, 722–733. [Google Scholar]
- British Electricity, I. CHAPTER 1—Nuclear physics and basic technology. In Nuclear Power Generation, 3rd ed.; Electricity, I.B., Ed.; Pergamon: Oxford, UK, 1992; pp. 1–110. [Google Scholar]
- de Oliviera, G.A.; Bustillos, J.O.; Ferreira, J.C.; Bergamaschi, V.S.; Moraes, R.M.D.; Gimenez, M.P.; Miyamoto, F.K.; Seneda, J.A. Applications of lithium in nuclear energy. In Proceedings of the INAC 2017: International Nuclear Atlantic Conference, Belo Horizonte, Brazil, 22–27 October 2017. [Google Scholar]
- Brooks, S.C.; Southworth, G.R. History of mercury use and environmental contamination at the Oak Ridge Y-12 Plant. Environ. Pollut. 2011, 159, 19–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.L.; Zhou, W.; Gu, L.; Qiu, D.; Ren, D.H.; Gu, Z.G.; Li, Z. Liquid-liquid extraction to lithium isotope separation based on room-temperature ionic liquids containing 2,2′-binaphthyldiyl-17-crown-5. J. Nucl. Sci. Technol. 2015, 52, 332–341. [Google Scholar] [CrossRef]
- Badea, S.-L.; Niculescu, V.-C.; Iordache, A.-M. New Trends in Separation Techniques of Lithium Isotopes: A Review of Chemical Separation Methods. Materials 2023, 16, 3817. [Google Scholar] [CrossRef]
- Klemm, A. Lithium in der Kerntechnik. Angew. Chem. 1958, 70, 21–24. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, P.; Ju, H.; Xue, Z.; Zhou, X.; Mao, L.; Shao, F.; Zou, X.; Jing, Y.; Jia, Y.; et al. Electromigration separation of lithium isotopes: The multiple roles of crown ethers. Chem. Phys. Lett. 2022, 787, 139265. [Google Scholar] [CrossRef]
- Hanada, T.; Goto, M. Synergistic Deep Eutectic Solvents for Lithium Extraction. ACS Sustain. Chem. Eng. 2021, 9, 2152–2160. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Zhu, A.; Bian, X.; Han, W.; Cao, D.; Wen, Y.; Zhu, K.; Wang, S. The application of deep eutectic solvents in lithium-ion battery recycling: A comprehensive review. Resour. Conserv. Recycl. 2023, 188, 106690. [Google Scholar] [CrossRef]
- Xie, Z.; Xie, M.; Tang, T.; Yang, F.; Xue, L.; Jiang, Z. β-Diketone-Driven Deep Eutectic Solvent for Ultra-Efficient Natural Stable Lithium-7 Isotope Separation. Separations 2023, 10, 111. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).