
A New Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry Method to Monitor 1-Amino-4-Methyl-Piperazine and 1-Methyl-4-Nitroso-Piperazine in Rifampicin Hydrolysis: A Standard Addition Approach to Quantify Nitrosamine in Rifampicin


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. LC-MS/MS Instrumentation and Conditions
2.3. Standard Solutions and Sample Preparation
2.4. Calibration Solutions
2.5. Method Validation Parameters for AMP and MNP Quantification in the RIF Hydrolysis Study
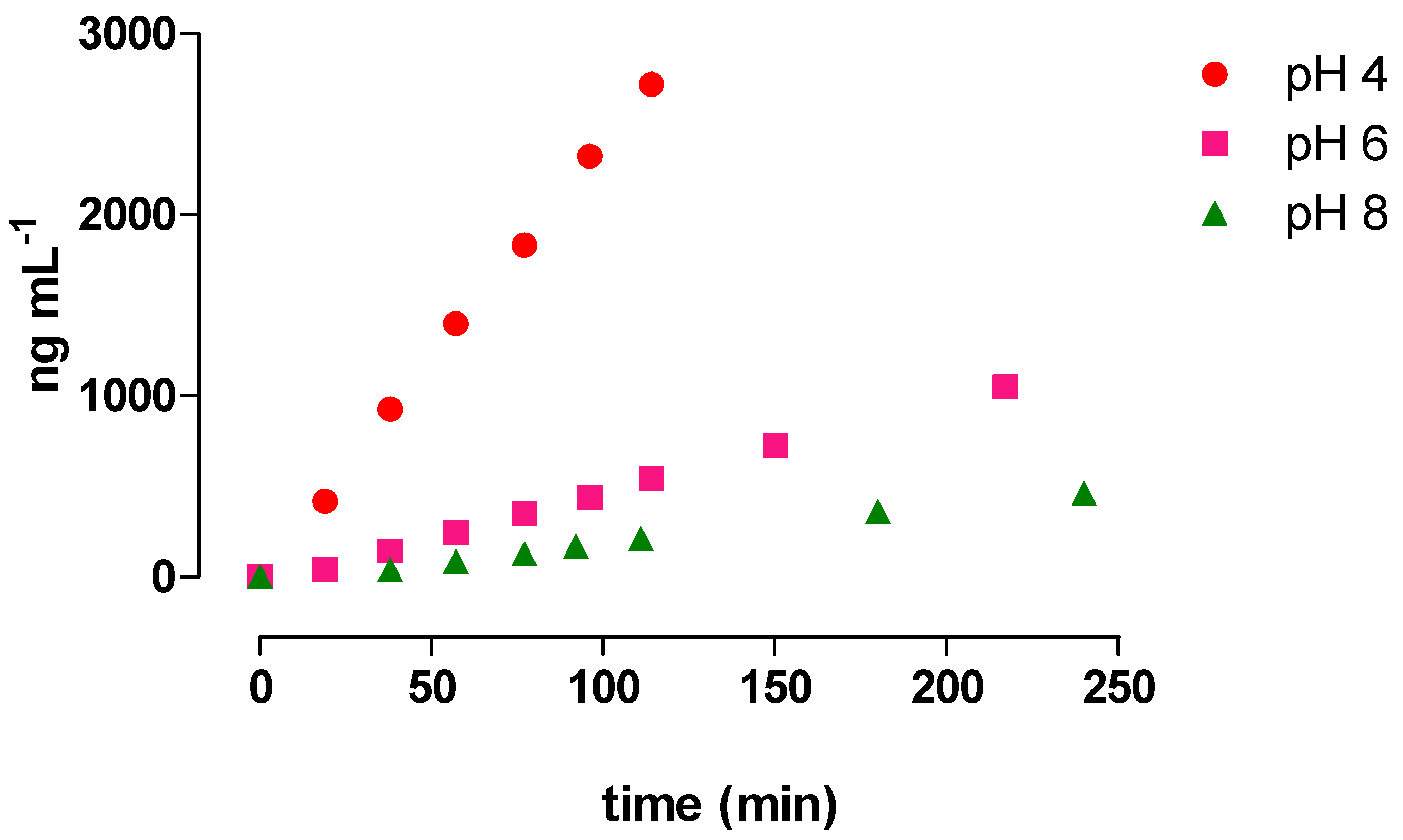
2.6. Monitoring of AMP and MNP Formation from RIF Hydrolysis in Buffered Solutions
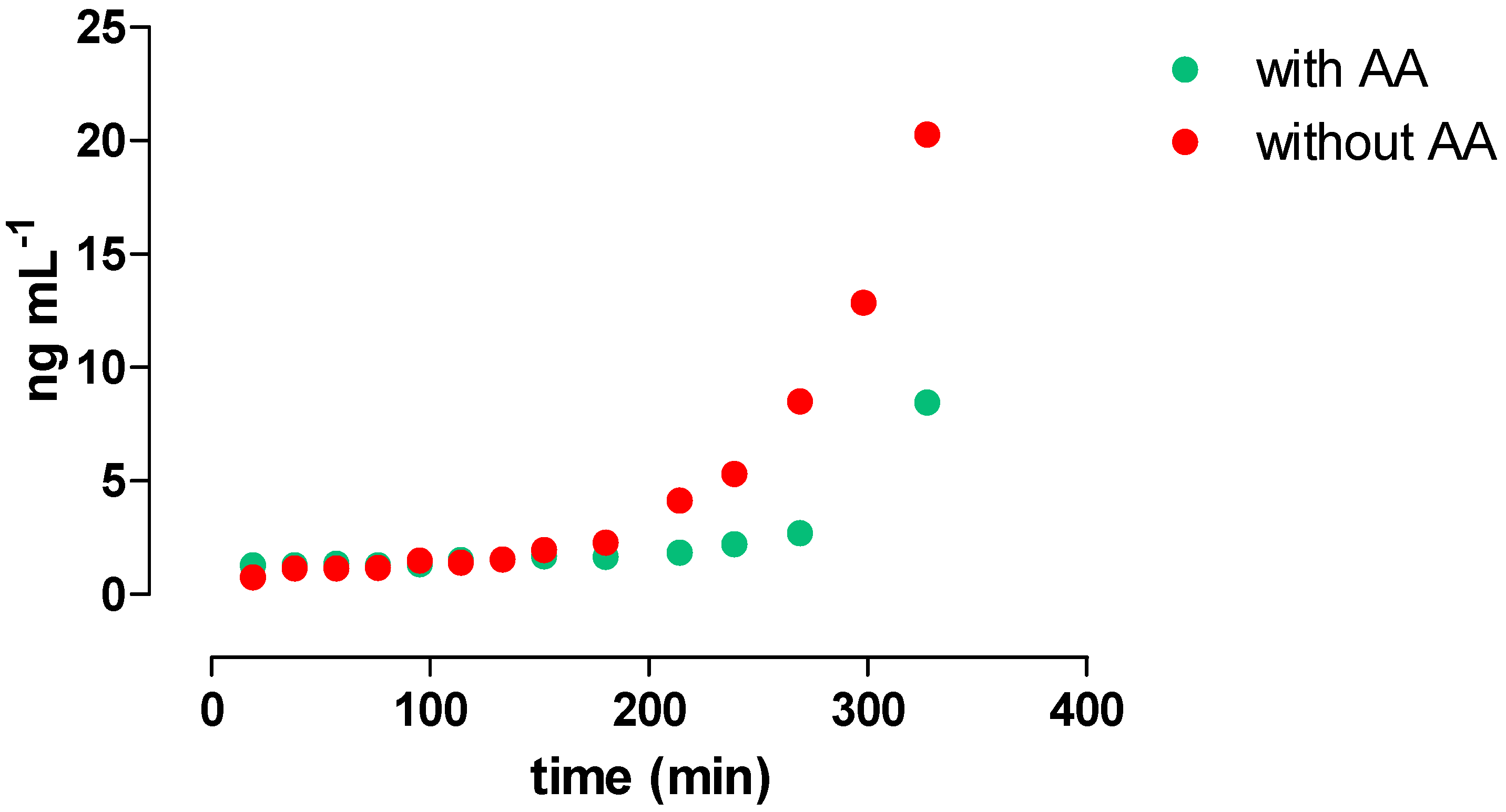
2.7. Monitoring of RIF Hydrolysis in Methanol
2.8. Standard Addition Method for Quantification of MNP in API
2.8.1. Sample Preparation
2.8.2. System Suitability Test
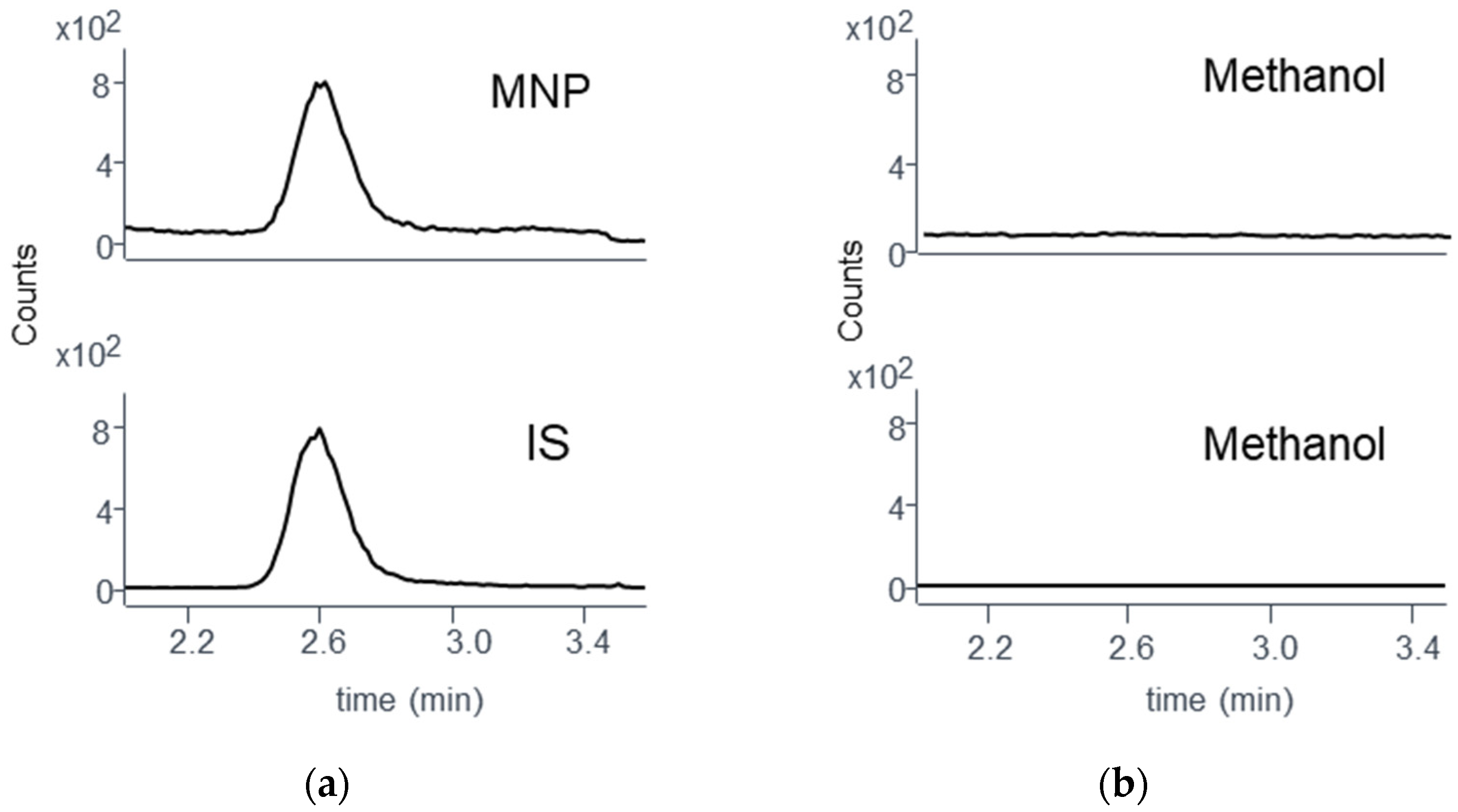
2.8.3. Specificity
2.8.4. Linearity
2.8.5. LOQ
2.8.6. Repeatability and Intermediate Precision
3. Results
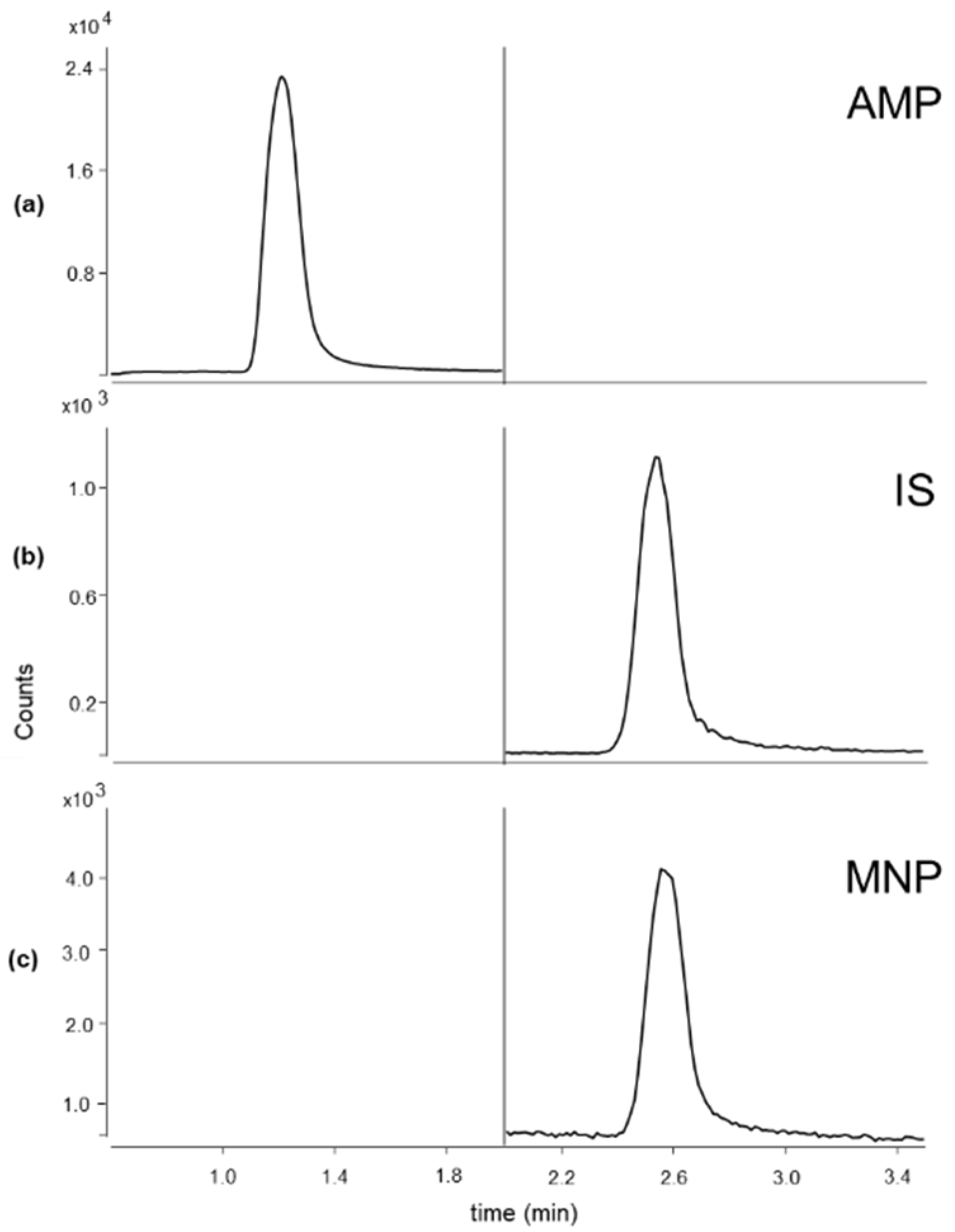
3.1. LC-MS/MS Conditions
3.2. Validation Results of the Method for the Quantification of AMP and MNP in the RIF Hydrolysis Study
3.3. Monitoring AMP and MNP Formation from RIF Hydrolysis in Buffered Solutions
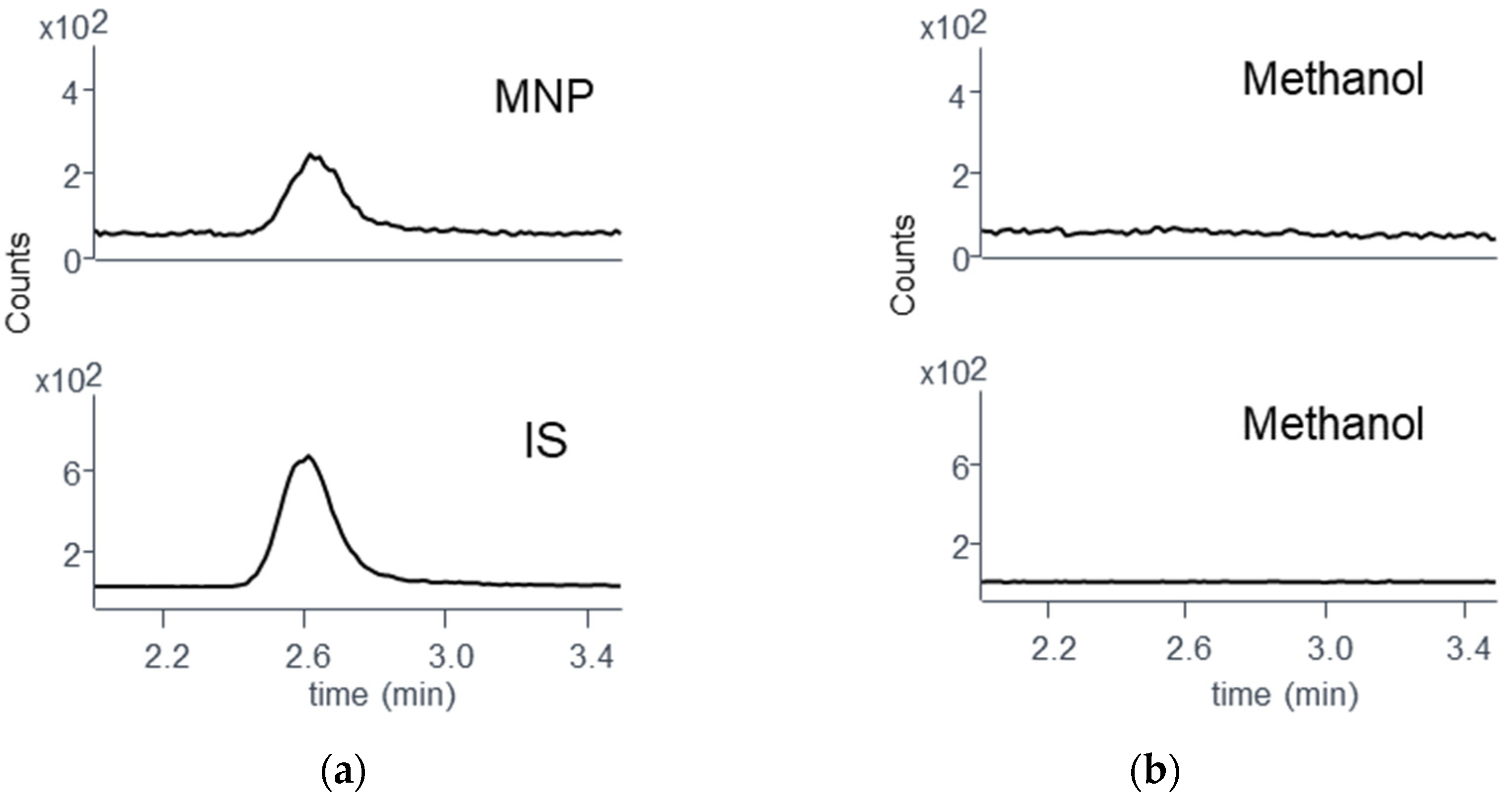
3.4. Monitoring RIF Hydrolysis in Methanol
3.5. Standard Addition Method for Quantitative Analysis of MNP in RIF
3.5.1. System Suitability Test
3.5.2. Specificity
3.5.3. Linearity
3.5.4. LOQ
3.5.5. Repeatability and Intermediate Precision
3.6. Comparison of the Two Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Samet, J.M.; Chiu, W.A.; Cogliano, V.; Jinot, J.; Kriebel, D.; Lunn, R.M.; Beland, F.A.; Bero, L.; Browne, P.; Fritschi, L.; et al. The IARC Monographs: Updated Procedures for Modern and Transparent Evidence Synthesis in Cancer Hazard Identification. J. Natl. Cancer Inst. 2020, 112, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Council of Europe. Substances for Pharmaceutical use (2034). In European Pharmacopoeia, 11th ed.; Supplement 11.3; EDQM: Strasbourg, France, 2022; Volume I, pp. 5085–5087. [Google Scholar]
- Council of Europe. Pharmaceutical Preparation (2619). In European Pharmacopoeia, 11th ed.; Supplement 11.3; EDQM: Strasbourg, France, 2022; Volume I, pp. 939–941. [Google Scholar]
- Nitrosamine impurities. Available online: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/referral-procedures/nitrosamine-impurities#guidance-for-marketing-authorisation-holders-section (accessed on 6 January 2024).
- Council of Europe. 2.5.42 N-Nitrosoamines in Active Pharmaceutical Ingredient. In European Pharmacopoeia, 11th ed.; Supplement 11.3; EDQM: Strasbourg, France, 2022; Volume I, pp. 199–203. [Google Scholar]
- Wohlfart, J.; Scherf-Clavel, O.; Kinzig, M.; Sörgel, F.; Holzgrabe, U. The nitrosamine contamination of drugs, part 3: Quantification of 4-Methyl-1-nitrosopiperazine in rifampicin capsules by LC-MS/HRMS. J. Pharm. Biomed. Anal. 2021, 203, 114205. [Google Scholar] [CrossRef] [PubMed]
- Seydel, J.K. Physico-chemical studies on rifampicin. Antibiot. Chemother. 1970, 16, 380–391. [Google Scholar] [PubMed]
- Ratti, B.; Parenti, R.; Toselli, A.; Zerilli, L. Quantitative assay of Rifampicin and its main metabolite 25-desacetylrifampicin in human plasma by reversed-phase high-performance liquid-chromatography. J. Chromatogr. 1981, 225, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Pranked, R.; Walters, J.; Parnes, J. Kinetics for degradation of Rifampicin, an azomethine-containing drug which exhibits reversible hydrolysis in acidic solutions. Int. J. Pharm. 1992, 78, 59–67. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Liquid Chromatography-High Resolution Mass Spectrometry (LC-ESI-HRMS) Method for the determination of MNP in Rifampin and CPNP in Rifapentine Drug Substance and Drug Product. 2020. Available online: https://www.fda.gov/media/142092/download (accessed on 6 January 2024).
- Tao, X.; Tian, Y.; Liu, W.; Yao, S.; Yin, L. Trace Level Quantification of 4-Methyl-1-nitrosopiperazin in Rifampicin Capsules by LC-MS/MS. Front. Chem. 2022, 10, 834124. [Google Scholar] [CrossRef] [PubMed]
- Witkowska, A.; Wolczynska, A.; Lis-Cieplak, A.; Stolarczyk, E. Development and Validation of LC-MS/MS Method for the Determination of 1-Methyl-4-Nitrosopiperazine (MNP) in Multicomponent Products with Rifampicin-Analytical Challenges and Degradation Studies. Molecules 2023, 28, 7405. [Google Scholar] [CrossRef] [PubMed]
- de Souza, G.; Matos, M.; Aglio, T.; Salles, A.J.; Rath, S. A comprehensive LC-UHPLC-MS/MS method for the monitoring of N-nitrosamines in lipophilic drugs: A case study with rifampicin. J. Pharm. Biomed. Anal. 2023, 236, 115685. [Google Scholar] [CrossRef] [PubMed]
- Shishoo, C.; Shah, S.; Rathod, I.; Savale, S.; Kotecha, J.; Shah, P. Stability of rifampicin in dissolution medium in presence of isoniazid. Int. J. Pharm. 1999, 190, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.S.; Murthy, K.V.R. Development of Dissolution Medium for Rifampicin Sustained Release Formulations. Indian J. Pharm. Sci. 2001, 63, 256–257. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RT | SRM1 | SRM2 | CE1/CE2 | Frag |
|---|---|---|---|---|---|
| AMP | 1.22 | 116.1 > 99.1 | 116.1 > 56.2 | 4/17 | 76 |
| MNP | 2.54 | 130.1 > 100.1 | 130.1 > 58.0 | 4/16 | 71 |
| IS | 2.56 | 134.1 > 104.1 | 134.1 > 58.2 | 4/20 | 71 |
| AMP | MNP | |
|---|---|---|
| AF | y = 1799x + 340,712 r2 = 0.998 | y = 431x − 68 r² = 0.999 |
| Methanol | y = 19,944x + 2249 r2 = 0.999 | y = 371x − 527 r² = 0.996 |
| Fortification Level (ng mL−1) | Recovery% | Repeatability | |
|---|---|---|---|
| 50 | 110 | 7 | |
| AMP | 500 | 97 | 5 |
| 2000 | 95 | 6 | |
| 25 | 119 | 8 | |
| MNP | 50 | 97 | 7 |
| 75 | 104 | 11 |
| Day | amean | bmean |
|---|---|---|
| 1 | 0.041 ± 0.001 | 0.870 ± 0.006 |
| 2 | 0.046 ± 0.001 | 0.941 ± 0.022 |
| 3 | 0.050 ± 0.001 | 0.964 ± 0.020 |
| Concentration (ng mL−1) | |||||
|---|---|---|---|---|---|
| Day | S1 | S2 | S3 | Smean | RSD% |
| 1 | 20.68 | 21.31 | 22.27 | 21.42 | 3.8 |
| 2 | 20.00 | 19.24 | 21.61 | 20.28 | 6.0 |
| 3 | 19.74 | 18.03 | 20.25 | 19.34 | 6.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panusa, A.; Rotundo, P.; Sadutto, D.; Rodomonte, A.; Cirilli, R. A New Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry Method to Monitor 1-Amino-4-Methyl-Piperazine and 1-Methyl-4-Nitroso-Piperazine in Rifampicin Hydrolysis: A Standard Addition Approach to Quantify Nitrosamine in Rifampicin. Separations 2024, 11, 54. https://doi.org/10.3390/separations11020054
Panusa A, Rotundo P, Sadutto D, Rodomonte A, Cirilli R. A New Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry Method to Monitor 1-Amino-4-Methyl-Piperazine and 1-Methyl-4-Nitroso-Piperazine in Rifampicin Hydrolysis: A Standard Addition Approach to Quantify Nitrosamine in Rifampicin. Separations. 2024; 11(2):54. https://doi.org/10.3390/separations11020054
Chicago/Turabian StylePanusa, Alessia, Paola Rotundo, Daniele Sadutto, Andrea Rodomonte, and Roberto Cirilli. 2024. "A New Ultra High-Performance Liquid Chromatography-Tandem Mass Spectrometry Method to Monitor 1-Amino-4-Methyl-Piperazine and 1-Methyl-4-Nitroso-Piperazine in Rifampicin Hydrolysis: A Standard Addition Approach to Quantify Nitrosamine in Rifampicin" Separations 11, no. 2: 54. https://doi.org/10.3390/separations11020054