Determination of Carminic Acid in Foodstuffs and Pharmaceuticals by Microchip Electrophoresis with Photometric Detection

 ,
, 
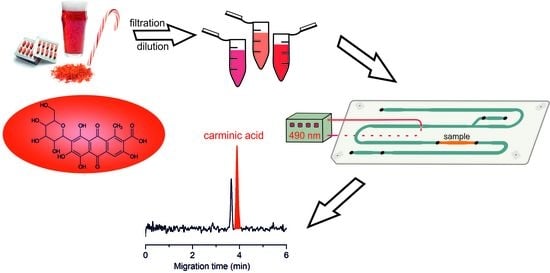
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Solutions
2.2. Food and Pharmaceutical Samples
2.3. MCE-PD Instrumentation
2.4. Microchip Maintenance
3. Results and Discussion
3.1. Separation Conditions
3.2. Method Performance Parameters
3.2.1. Linearity
3.2.2. Limit of Detection and Limit of Quantitation
3.2.3. Precision
3.2.4. Accuracy
3.3. Analysis of Food and Pharmaceutical Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BGE | Background electrolyte |
| CA | Carminic acid |
| CE | Capillary electrophoresis |
| EFSA | European Food Safety Authority |
| FAO | Food and Agriculture Organization of the United Nations |
| HIS | L-histidine |
| HPLC | High performance liquid chromatography |
| HVPS | High voltage power supply |
| HVR | High voltage relay |
| i.d. | Internal diameter |
| LDR | Linear dynamic range |
| LOD | Limit of detection |
| LOQ | Limit of quantitation |
| MCE | Microchip electrophoresis |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| MHEC | Methylhydroxyethylcellulose |
| MPL | Maximum permitted level |
| PD | Photometric detection |
| RSD | Relative standard deviation |
| SD | Standard deviation |
| UV–Vis | Ultraviolet-visible |
| WHO | World Health Organization |
References
- Amchova, P.; Kotolova, H.; Ruda-Kucerova, J. Health safety issues of synthetic food colorants. Regul. Toxicol. Pharmacol. 2015, 73, 914–922. [Google Scholar] [CrossRef]
- Scotter, M.J. Colour Additives for Food and Beverages; Woodhead Publishing: Cambridge, UK, 2015. [Google Scholar]
- The European Parliament and the Council of the European Union. Council Regulation (EC) 1333/2008 of 16 December 2008 on food additives. Off. J. Eur. Union 2008, L354, 16–33. [Google Scholar]
- The European Parliament and the Council of the European Union. Directive 2009/35/EC of the European Parliament and of the Council of 23 April 2009 on Colouring Matters Which May Be Added to Medical Products. Off. J. Eur. Union 2009, L109, 10–13. [Google Scholar]
- Martins, N.; Roriz, C.L.; Morales, P.; Barros, L.; Ferreira, I.C.F.R. Food colorants: Challenges, opportunities and current desires of agro-industries to ensure consumer expectations and regulatory practices. Trends Food Sci. Technol. 2016, 52, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Vargas, F.; Paredes-López, O. Natural Colorants for Food and Nutraceutical Uses; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Samari, F.; Hemmateenejad, B.; Shamsipur, M. Spectrophotometric determination of carminic acid in human plasma and fruit juices by second order calibration of the absorbance spectra–pH data matrices coupled with standard addition method. Anal. Chim. Acta 2010, 667, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Merinas-Amo, R.; Martínez-Jurado, M.; Jurado-Güeto, S.; Alonso-Moraga, Á.; Merinas-Amo, T. Biological effects of food coloring in in vivo and in vitro model systems. Foods 2019, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA Panel on Food Additives and Nutrient Sources added to Food. Scientific Opinion on the re-evaluation of cochineal, carminic acid, carmines (E 120) as a food additive. EFSA J. 2015, 13. [Google Scholar] [CrossRef]
- Scotter, M.J. Methods for the determination of European Union-permitted added natural colours in foods: A review. Food Addit. Contam. Part A Chem. 2011, 28, 527–596. [Google Scholar] [CrossRef] [PubMed]
- Ntrallou, K.; Gika, H.; Tsochatzis, E. Analytical and sample preparation techniques for the determination of food colorants in food matrices. Foods 2020, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- The European Commission. Commission Regulation (EU) No 231/2012 of 9 March 2012 laying down specifications for food additives listed in Annexes II and III to Regulation (EC) No 1333/2008 of the European Parliament and of the Council. Off. J. Eur. Union 2012, L83, 1–295. [Google Scholar]
- Heydari, R.; Hosseini, M.; Zarabi, S. A simple method for determination of carmine in food samples based on cloud point extraction and spectrophotometric detection. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 150, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.H.; Alshammery, H.M.; Abdalla, M.A.; Alghamdi, A.F. Determination of carmine food dye (E120) in foodstuffs by stripping voltammetry. J. AOAC Int. 2009, 92, 1454–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, U.T.; Ergun, F.; Yilmaz, H. Determination of the food dye carmine in milk and candy products by differential pulse polarography. J. Food Drug Anal. 2014, 22, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šuleková, M.; Hudák, A.; Smřcová, M. The determination of food dyes in vitamins by RP-HPLC. Molecules 2016, 21, 1368. [Google Scholar] [CrossRef]
- Lim, H.S.; Choi, J.C.; Song, S.B.; Kim, M. Quantitative determination of carmine in foods by high-performance liquid chromatography. Food Chem. 2014, 158, 521–526. [Google Scholar] [CrossRef]
- Ordoudi, S.A.; Staikidou, C.; Kyriakoudi, A.; Tsimidou, M.Z. A stepwise approach for the detection of carminic acid in saffron with regard to religious food certification. Food Chem. 2018, 267, 410–419. [Google Scholar] [CrossRef]
- Carvalho, P.R.N.; Collins, C.H. HPLC determination of carminic acid in foodstuffs and beverages using diode array and fluorescence detection. Chromatographia 1997, 45, 63–66. [Google Scholar] [CrossRef]
- Feng, F.; Zhao, Y.; Yong, W.; Sun, L.; Jiang, G.; Chu, X. Highly sensitive and accurate screening of 40 dyes in soft drinks by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1813–1818. [Google Scholar] [CrossRef]
- Huang, H.Y.; Chiu, C.W.; Sue, S.L.; Cheng, C.F. Analysis of food colorants by capillary electrophoresis with large-volume sample stacking. J. Chromatogr. A 2003, 995, 29–36. [Google Scholar] [CrossRef]
- Nevado, J.J.B.; Cabanillas, C.G.; Salcedo, A.M.C. Method development and validation for the simultaneous determination of dyes in foodstuffs by capillary zone electrophoresis. Anal. Chim. Acta 1999, 378, 63–71. [Google Scholar] [CrossRef]
- Huang, H.Y.; Shih, Y.C.; Chen, Y.C. Determining eight colorants in milk beverages by capillary electrophoresis. J. Chromatogr. A 2002, 959, 317–325. [Google Scholar] [CrossRef]
- Huang, H.Y.; Chuang, C.L.; Chiu, C.W.; Chung, M.C. Determination of food colorants by microemulsion electrokinetic chromatography. Electrophoresis 2005, 26, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-J.; Liu, C.-T.; Li, W.; Tang, A.-N. Dispersive solid-phase microextraction and capillary electrophoresis separation of food colorants in beverages using diamino moiety functionalized silica nanoparticles as both extractant and pseudostationary phase. Talanta 2015, 132, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Musso, N.; Grasso, M.; Costantino, A.; Lazzarino, G.; Tascedda, F.; Gulisano, M.; Lunte, S.M.; Caraci, F. Microfluidics as a novel tool for biological and toxicological assays in drug discovery processes: Focus on microchip electrophoresis. Micromachines 2020, 11, 593. [Google Scholar] [CrossRef]
- Guzman, N.A.; Guzman, D.E. A two-dimensional affinity capture and separation mini-platform for the isolation, enrichment, and quantification of biomarkers and its potential use for liquid biopsy. Biomedicines 2020, 8, 255. [Google Scholar] [CrossRef]
- Kaniansky, D.; Masár, M.; Bodor, R.; Žúborová, M.; Ölvecká, M.; Jöhnck, M.; Stanislawski, B. Electrophoretic separations on chips with hydrodynamically closed separation systems. Electrophoresis 2003, 24, 2208–2227. [Google Scholar] [CrossRef]
- Rasimas, J.P.; Berglund, K.A.; Blanchard, G.J. A molecular lock-and-key approach to detecting solution phase self-assembly. A fluorescence and absorption study of carminic acid in aqueous glucose solutions. J. Phys. Chem. 1996, 100, 7220–7229. [Google Scholar] [CrossRef]
- International Conference on Harmonisation of Technical Requirements of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 20 October 2020).
- ISO. International Standard ISO 3632-2: Spices—Saffron (Crocus sativus L.). Part 2: Test methods; ISO: Geneva, Switzerland, 2010. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Mean noise ± SD | 0.53 ± 0.13 mV |
| Equation of regression line 1 | y = 6.204x + 0.376 |
| Correlation coefficient 1 | 0.9972 |
| Calibration range 1 | 0.2–1.0 µmol L−1 |
| LOD 2 | 69 nmol L−1 |
| LOQ 3 | 207 nmol L−1 |
| Concentration (µmol L−1) | Intra-Day Precision 1 (n = 4) | Inter-Day Precision 1 (n = 20) | ||
|---|---|---|---|---|
| Migration Time | Peak Area | Migration Time | Peak Area | |
| 0.2 | 0.75 | 1.13 | 1.38 | 1.88 |
| 1.0 | 1.18 | 1.22 | 1.86 | 1.20 |
| 5.0 | 0.97 | 1.12 | 2.45 | 1.38 |
| Sample | Added Concentration (µmol L−1) | Accuracy (n = 4) | Dilution Factor |
|---|---|---|---|
| Mean ± SD (%) | |||
| Model sample | 0.2 | 98.52 ± 1.11 | |
| 1.0 | 98.42 ± 1.20 | ||
| 5.0 | 100.72 ± 1.13 | ||
| Soft candy | 0.50 | 94.53 ± 2.01 | 5 |
| 1.0 | 95.36 ± 1.20 | ||
| Hard candy | 0.30 | 99.24 ± 2.39 | 40 |
| 0.45 | 103.08 ± 1.46 | ||
| Radler | 0.30 | 98.05 ± 2.61 | 2 |
| 0.45 | 97.95 ± 2.75 | ||
| Saffron | 0.50 | 99.22 ± 1.41 | 2 |
| 1.0 | 92.75 ± 2.26 | ||
| Medicinal lollipops | 0.25 | 92.50 ± 2.11 | 40 |
| 0.75 | 93.70 ± 0.89 | ||
| Lozenges | 0.30 | 103.96 ± 1.09 | 2.5 |
| 0.45 | 92.70 ± 1.61 |
| Sample | Added Concentration (µmol L−1) | Intra-Day Precision 1 (n = 4) | Dilution Factor | |
|---|---|---|---|---|
| Migration Time | Peak Area | |||
| Soft candy | 0 | 1.06 | 1.37 | 5 |
| 0.50 | 1.18 | 0.74 | ||
| 1.0 | 0.41 | 0.65 | ||
| Hard candy | 0 | 1.15 | 1.88 | 40 |
| 0.30 | 1.15 | 1.07 | ||
| 0.45 | 0.77 | 0.79 | ||
| Radler | 0 | 1.24 | <LOQ | 2 |
| 0.30 | 1.08 | 1.53 | ||
| 0.45 | 0.92 | 1.88 | ||
| Saffron | 0 | 0.60 | 1.88 | 2 |
| 0.50 | 0.38 | 0.54 | ||
| 1.0 | 0.50 | 1.31 | ||
| Medicinal lollipops | 0 | 0.55 | 1.49 | 40 |
| 0.25 | 0.27 | 0.78 | ||
| 0.75 | 1.08 | 0.58 | ||
| Lozenges | 0 | 0.41 | 1.27 | 2.5 |
| 0.30 | 0.28 | 0.49 | ||
| 0.45 | 0.60 | 0.94 | ||
| Sample | Concentration of CA ± SD (µmol L−1) | Content of CA (mg kg−1) | MPL 1 (mg kg−1) |
|---|---|---|---|
| Soft candy | 4.43 ± 0.06 | 7.85 ± 0.11 | 300 |
| Hard candy | 15.07 ± 0.28 | 8.73 ± 0.16 | 300 |
| Radler | <LOQ | <LOQ | 200 2 |
| Saffron | 1.59 ± 0.03 | 78.45 ± 1.47 | 500 |
| Medicinal lollipops | 17.82 ± 0.26 | 35.00 ± 0.52 | 300 3 |
| Lozenges | 0.88 ± 0.01 | 3.41 ± 0.04 | 300 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masár, M.; Hradski, J.; Vargová, E.; Miškovčíková, A.; Božek, P.; Ševčík, J.; Szucs, R. Determination of Carminic Acid in Foodstuffs and Pharmaceuticals by Microchip Electrophoresis with Photometric Detection. Separations 2020, 7, 72. https://doi.org/10.3390/separations7040072
Masár M, Hradski J, Vargová E, Miškovčíková A, Božek P, Ševčík J, Szucs R. Determination of Carminic Acid in Foodstuffs and Pharmaceuticals by Microchip Electrophoresis with Photometric Detection. Separations. 2020; 7(4):72. https://doi.org/10.3390/separations7040072
Chicago/Turabian StyleMasár, Marián, Jasna Hradski, Eva Vargová, Adriána Miškovčíková, Peter Božek, Juraj Ševčík, and Roman Szucs. 2020. "Determination of Carminic Acid in Foodstuffs and Pharmaceuticals by Microchip Electrophoresis with Photometric Detection" Separations 7, no. 4: 72. https://doi.org/10.3390/separations7040072
APA StyleMasár, M., Hradski, J., Vargová, E., Miškovčíková, A., Božek, P., Ševčík, J., & Szucs, R. (2020). Determination of Carminic Acid in Foodstuffs and Pharmaceuticals by Microchip Electrophoresis with Photometric Detection. Separations, 7(4), 72. https://doi.org/10.3390/separations7040072