
Pattern-Type Separation of Triacylglycerols by Silver Thiolate×Non-Aqueous Reversed Phase Comprehensive Liquid Chromatography


Abstract
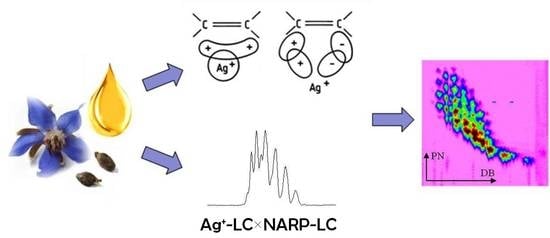
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Sample Preparation
2.2. Instruments and Analytical Conditions
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donato, P.; Cacciola, F.; Beccaria, M.; Dugo, P.; Mondello, L. Lipidomics. In Comprehensive Analytical Chemistry, Advanced Mass Spectrometry for Food Safety and Quality; Picó, Y., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 68, pp. 395–439. [Google Scholar]
- Buchgraber, M.; Franz, U.; Hendrik, E.; Elke, A. Triacylglycerol profiling by using chromatographic techniques. Eur. J. Lipid Sci. Technol. 2004, 106, 621–648. [Google Scholar] [CrossRef]
- Donato, P.; Dugo, P.; Mondello, L. Separation of Lipids. In Liquid Chromatography; Fanali, S., Haddad, P.R., Poole, C.F., Schoenmakers, P., Lloyd, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 203–248. [Google Scholar]
- Donato, P.; Inferrera, V.; Sciarrone, D.; Mondello, L. Supercritical fluid chromatography for lipid analysis in foodstuffs. J. Sep. Sci. 2017, 40, 361–382. [Google Scholar] [CrossRef]
- Donato, P.; Micalizzi, G.; Oteri, M.; Rigano, F.; Sciarrone, D.; Dugo, P.; Mondello, L. Comprehensive lipid profiling in the mediterranean mussel (Mytilus galloprovincialis) using hyphenated and multidimensional chromatography techniques coupled to mass spectrometry detection. Anal. Bioanal. Chem. 2018, 410, 3297–3313. [Google Scholar] [CrossRef]
- Holčapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef] [Green Version]
- Holčapek, M.; Lísa, M. Silver-ion liquid chromatography–mass spectrometry. In Handbook of Advanced Chromatography/Mass Spectrometry Techniques; Holčapek, M., Byrdwell, W.C., Eds.; Elsevier: London, UK, 2017; Volume 3, pp. 115–140. [Google Scholar]
- Dobson, G.; Christie, W.W.; Nikolova-Damyanova, B. Silver ion chromatography of lipids and fatty acids. J. Chromatogr. B Biomed. Appl. 1995, 671, 197–222. [Google Scholar] [CrossRef]
- Momchilova, S.M.; Nikolova-Damyanova, B.M. Advances in silver ion chromatography for the analysis of fatty acids and triacylglycerols-2001 to 2011. Anal. Sci. 2012, 28, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, J.T.; Aponte, J.C.; Tarozo, R.; Huang, Y. Efficient liquid chromatographic analysis of mono-, di-, and triglycerols using silver thiolate stationary phase. J. Chromatogr. A 2012, 1240, 90–95. [Google Scholar] [CrossRef]
- Aponte, J.C.; Dillon, J.T.; Tarozo, R.; Huang, Y. Separation of unsaturated organic compounds using silver–thiolate chromatographic material. J. Chromatogr. A 2012, 1240, 83–89. [Google Scholar] [CrossRef]
- Cacciola, F.; Donato, P.; Sciarrone, D.; Dugo, P.; Mondello, L. Comprehensive liquid chromatography and other liquid-based comprehensive techniques coupled to mass spectrometry in food analysis. Anal. Chem. 2017, 89, 414–429. [Google Scholar] [CrossRef]
- Dugo, P.; Beccaria, M.; Fawzy, N.; Donato, P.; Cacciola, F.; Mondello, L. Mass spectrometric elucidation of triacylglycerol content of Brevoortia tyrannus (menhaden) oil using non-aqueous reversed-phase liquid chromatography under ultra high pressure conditions. J. Chromatogr. A 2012, 1259, 227–236. [Google Scholar] [CrossRef]
- Lísa, M.; Holčapek, M. Triacylglycerols in nut and seed oils: Detailed characterization using high-performance liquid chromatography/mass spectrometry. In Nuts and Seeds in Health and Disease Prevention; Preedy, V.R., Watson, R.R., Patel, V.B., Eds.; Academic Press: London, UK, 2011; pp. 43–54. [Google Scholar]
- Holčapek, M.; Velínská, H.; Lísa, M.; Cesla, P. Orthogonality of silver-ion and non-aqueous reversed-phase HPLC/MS in the analysis of complex natural mixtures of triacylglycerols. J. Sep. Sci. 2009, 32, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Stoll, D.R.; Carr, P.W. Two-Dimensional liquid chromatography: A state of the art tutorial. Anal. Chem. 2017, 89, 519–531. [Google Scholar] [CrossRef]
- Mondello, L.; Beccaria, M.; Donato, P.; Cacciola, F.; Dugo, G.; Dugo, P. Comprehensive two-dimensional liquid chromatography with evaporative light scattering detection for the analysis of triacylglycerols in Borago officinalis. J. Sep. Sci. 2011, 34, 688–692. [Google Scholar] [CrossRef]
- Holčapek, M.; Lísa, M.; Jandera, P.; Kabátová, N. Quantitation of triacylglycerols in plant oils using HPLC with APCI-MS, evaporative light-scattering, and UV detection. J. Sep. Sci. 2005, 28, 1315–1333. [Google Scholar] [CrossRef]
- Holčapek, M.; Jandera, P.; Zderadička, P.; Hrubá, L. Characterization of triacylglycerol and diacylglycerol composition of plant oils using high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 2003, 1010, 195–215. [Google Scholar] [CrossRef]
- Eskin, N.A.M. Borage and evening primrose oil. Eur. J. Lipid Sci. Technol. 2008, 110, 651–654. [Google Scholar] [CrossRef]
- Asadi-Samani, M.; Bahmani, M.; Rafieian-Kopaei, M. The chemical composition, botanical characteristic and biological activities of Borago officinalis: A review. Asian Pac. J. Trop. Med. 2014, 7, S22–S28. [Google Scholar] [CrossRef] [Green Version]
- Lísa, M.; Holčapek, M.; Boháč, M. Statistical Evaluation of Triacylglycerol Composition in Plant Oils Based on High-Performance Liquid Chromatography Atmospheric Pressure Chemical Ionization Mass Spectrometry Data. J. Agric. Food Chem. 2009, 57, 6888–6898. [Google Scholar] [CrossRef]
- Lísa, M.; Holčapek, M. Triacylglycerols profiling in plant oils important in food industry, dietetics and cosmetics using high-performance liquid chromatography–atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 2008, 1198–1199, 115–130. [Google Scholar] [CrossRef]
- Ismail, O.H.; Catani, M.; Pasti, L.; Cavazzini, A.; Ciogli, A.; Villani, C.; Kotoni, D.; Gasparrini, F.; Bell, D.S. Experimental evidence of the kinetic performance achievable with columns packed with the new 1.9 μm fully porous particles Titan C18. J. Chromatogr. A 2016, 1454, 86–92. [Google Scholar] [CrossRef]
- Sommella, E.; Pepe, G.; Ventre, G.; Pagano, F.; Manfra, M.; Pierri, G.; Ismail, O.; Ciogli, A.; Campiglia, P. Evaluation of two sub-2 μm stationary phases, core-shell and totally porous monodisperse, in the second dimension of on-line comprehensive two dimensional liquid chromatography, a case study: Separation of milk peptides after expiration date. J. Chromatogr. A 2015, 1375, 54–61. [Google Scholar] [CrossRef]
- Broeckhoven, K.; Desmet, G. Advances and Innovations in Liquid Chromatography Stationary Phase Supports. Anal. Chem. 2021, 93, 257–272. [Google Scholar] [CrossRef]
- Catani, M.; Ismail, O.H.; Cavazzini, A.; Ciogli, A.; Villani, C.; Pasti, L.; Bergantin, C.; Cabooter, D.; Desmet, G.; Gasparrini, F.; et al. Rationale behind the optimum efficiency of columns packed with new 1.9 µm fully porous particles of narrow particle size distribution. J. Chromatogr. A 2016, 1454, 78–85. [Google Scholar] [CrossRef]
- Neue, U.D. Theory of peak capacity in gradient elution. J. Chromatogr. A 2005, 1079, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Horváth, K.; Fairchild, J.N.; Guiochon, G. Generation and Limitations of Peak Capacity in Online Two-Dimensional Liquid Chromatography. Anal. Chem. 2009, 81, 3879–3888. [Google Scholar] [CrossRef]
- Gu, H.; Huang, Y.; Carr, P.W. Peak capacity optimization in comprehensive two dimensional liquid chromatography: A practical approach. J. Chromatogr. A 2011, 1218, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.M.; Stoll, D.R.; Carr, P.W. Effect of First-Dimension Undersampling on Effective Peak Capacity in Comprehensive Two-Dimensional Separations. Anal. Chem. 2008, 80, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Lísa, M.; Netušilová, K.; Franěk, L.; Dvořáková, H.; Vrkoslav, V.; Holčapek, M. Characterization of fatty acid and triacylglycerol composition in animal fats using silver-ion and non-aqueous reversed-phase high-performance liquid chromatography/mass spectrometry and gas chromatography/flame ionization detection. J. Chromatogr. A 2011, 1218, 7499–7510. [Google Scholar] [CrossRef]
- Nikolova-Damyanova, B. Retention of lipids in silver ion high-performance liquid chromatography: Facts and assumptions. J. Chromatogr. A 2009, 1216, 1815–1824. [Google Scholar] [CrossRef]
- Christie, W.W. Separation of molecular species of triacylglycerols by high-performance liquid chromatography with a silver ion column. J. Chromatogr. A 1988, 454, 273–284. [Google Scholar] [CrossRef]
- Neff, W.E.; Adlof, R.O.; List, G.R.; El-Agaimy, M. Analyses of Vegetable Oil Triacylglycerols by Silver Ion High Performance Liquid Chromatography with Flame Ionization Detection. J. Liq. Chromatogr. 1994, 17, 3951–3968. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Retention Time (Minutes) | |||
|---|---|---|---|---|
| Intra-Day Assays | Inter-Day Assays | |||
| 1 | 16.19 ± 0.05 | 0.30 | 16.29 ± 0.09 | 0.55 |
| 2 | 17.65 ± 0.03 | 0.17 | 17.72 ± 0.09 | 0.51 |
| 3 | 19.12 ± 0.05 | 0.26 | 19.19 ± 0.08 | 0.42 |
| 4 | 20.59 ± 0.06 | 0.29 | 20.72 ± 0.20 | 0.97 |
| 5 | 22.47 ± 0.06 | 0.27 | 22.52 ± 0.08 | 0.36 |
| 6 | 24.49 ± 0.06 | 0.24 | 24.53 ± 0.08 | 0.33 |
| 7 | 26.53 ± 0.05 | 0.19 | 26.57 ± 0.07 | 0.26 |
| 8 | 28.50 ± 0.04 | 0.14 | 28.51 ± 0.07 | 0.25 |
| Peak | Retention Time (Minutes) | Peak Area | ||
|---|---|---|---|---|
| 1 | 141.005 ± 1.23 | 0.92 | 37,513,401 ± 2,479,635.81 | 6.61 |
| 2 | 111.005 ± 2.01 | 1.81 | 712,277,629 ± 40,314,913.8 | 5.66 |
| 3 | 97.505 ± 0.84 | 0.86 | 677,656,664 ± 45,470,762.2 | 6.71 |
| 4 | 73.505 ± 0.75 | 1.02 | 483,139,425 ± 29,906,330.4 | 6.19 |
| 5 | 61.505 ± 0.20 | 0.33 | 9,674,556 ± 248,636.089 | 2.57 |
| # | TAG | DB | PN | [M+H]+ Theor | [M+H]+ Exp | DG+ Exp | DG+ Exp | DG+ Exp | Error (ppm) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PPP | 0 | 48 | 807.7436 | nd | PP, 551.4641 | - | ||
| 2 | SPP | 0 | 50 | 835.7749 | nd | SP, 579.5520 | PP, 551.4970 | - | |
| 3 | POP | 1 | 48 | 833.7593 | nd | PP, 551.4877 | PO, 577.5152 | - | |
| 4 | SOP | 1 | 50 | 861.7906 | nd | OP, 577.5122 | SP, 579.5578 | SO, 605.5433 | - |
| 5 | SOS | 1 | 52 | 889.8219 | nd | SO, 605.5468 | SS, 607.5575 | - | |
| 6 | AOP | 1 | 52 | 889.8219 | nd | AO, 633.5779 | OP, 577.5107 | AP, 607.5575 | - |
| 7 | BOP | 1 | 54 | 917.8532 | nd | BO, 661.5996 | OP, 577.5149 | BP, 635.5797 | - |
| 8 | AOS | 1 | 54 | 917.8532 | nd | AO, 633.5785 | OS, 605.5499 | AS, 635.5797 | - |
| 9 | LgOP | 1 | 56 | 945.8845 | nd | LgO, 689.6104 | OP, 577.5140 | LgP, 663.6274 | - |
| 10 | BOS | 1 | 56 | 945.8845 | nd | BO, 661.6147 | SO, 605.5388 | BS, 663.62840 | - |
| 11 | PLP | 2 | 46 | 831.7436 | 831.7494 | PP, 551.5020 | PL, 575.4978 | 6.9 | |
| 12 | OOP | 2 | 48 | 859.7749 | 859.7733 | PO, 577.5152 | OO, 603.5265 | 1.9 | |
| 13 | SLP | 2 | 48 | 859.7749 | 859.7733 | SL, 603.5265 | LP, 575.4973 | SL, 603.5265 | 1.9 |
| 14 | GOP | 2 | 50 | 887.8062 | 887.8080 | GO, 631.5566 | OP, 577.5161 | GP, 605.5420 | 2.0 |
| 15 | ALP | 2 | 50 | 887.8062 | 887.8080 | AL, 631.5565 | LP, 575.505 | AP, 607.5447 | 2.0 |
| 16 | SLS | 2 | 50 | 887.8062 | 887.8080 | SL, 603.5271 | SS, 607.5447 | 2.0 | |
| 17 | SOO | 2 | 50 | 887.8062 | 887.8080 | OO, 603.5271 | SO, 605.5420 | 2.0 | |
| 18 | C22:1OP | 2 | 52 | 915.8375 | 915.8354 | C22:1O, 659.5895 | OP, 577.5151 | C22:1P, 633.5715 | 2.3 |
| 19 | AOO | 2 | 52 | 915.8375 | 915.8354 | AO, 633.5715 | OO, 603.5296 | 2.3 | |
| 20 | GOS | 2 | 52 | 915.8375 | 915.8354 | GO, 631.5572 | SO, 605.5437 | GS, 633.5715 | 2.3 |
| 21 | BLP | 2 | 52 | 915.8375 | 915.8354 | BL, 659.5895 | LP, 575.4986 | BP, 635.6066 | 2.3 |
| 22 | ALS | 2 | 52 | 915.8375 | 915.8354 | AL, 631.5572 | LO, 601.5243 | AO, 633.5715 | 2.3 |
| 23 | C24:1OP | 2 | 54 | 943.8688 | 943.8705 | C24:1O, 687.6199 | OP, 577.5144 | C24:1P, 661.6041 | 1.8 |
| 24 | C22:1OS | 2 | 54 | 943.8688 | 943.8705 | C22:1O, 659.5888 | OS, 605.5433 | C22:1S, 661.6041 | 1.8 |
| 25 | BOO | 2 | 54 | 943.8688 | 943.8705 | BO, 661.6041 | OO, 603.5324 | 1.8 | |
| 26 | LgLP | 2 | 54 | 943.8688 | 943.8705 | LgL, 687.6196 | LP, 575.4772 | LgP, 663.6432 | 1.8 |
| 27 | BLS | 2 | 54 | 943.8688 | 943.8705 | BL, 659.5888 | LS, 603.5324 | BS, 663.6432 | 1.8 |
| 28 | C24:1OS | 2 | 56 | 971.9001 | nd | C24:1O, 687.5943 | OS, 605.5515 | C24:1S, 689.6465 | - |
| 29 | LgOO | 2 | 56 | 971.9001 | nd | LgO, 689.6465 | OO, 603.4264 | - | |
| 30 | LgLS | 2 | 56 | 971.9001 | nd | LgL, 687.5943 | LS, 603.4264 | LgS, 691.6627 | - |
| 31 | PγLnP | 3 | 44 | 829.7280 | 829.7257 | PP, 551.5008 | PγLn, 573.4831 | 2.8 | |
| 32 | OLP | 3 | 46 | 857.7593 | 857.7545 | PL, 575.5003 | PO, 577.5140 | LO, 601.5109 | 5.6 |
| 33 | SγLnP | 3 | 46 | 857.7593 | 857.7545 | SγLn, 601.5109 | PγLn, 573.4830 | SP, 579.5282 | 5.6 |
| 34 | OOO | 3 | 48 | 885.7906 | 885.7939 | OO, 603.5292 | 3.7 | ||
| 35 | GLP | 3 | 48 | 885.7906 | 885.7939 | GL, 629.5392 | LP, 575.5004 | GP, 605.5375 | 3.7 |
| 36 | SLO | 3 | 48 | 885.7906 | 885.7939 | LO, 601.5169 | SL, 603.5292 | SO, 605.5375 | 3.7 |
| 37 | SγLnS | 3 | 48 | 885.7906 | 885.7939 | SS, 607.5575 | SγLn, 601.5169 | 3.7 | |
| 38 | ALO | 3 | 50 | 913.8219 | 913.8202 | AL, 631.5587 | LO, 601.5143 | AO, 633.5729 | 1.9 |
| 39 | BγLnP | 3 | 50 | 913.8219 | 913.8202 | BγLn, 657.5726 | PγLn, 573.4856 | BP, 635.5901 | 1.9 |
| 40 | GLS | 3 | 50 | 913.8219 | 913.8202 | GL, 629.5421 | LS, 603.5285 | GS, 633.5729 | 1.9 |
| 41 | GOO | 3 | 50 | 913.8219 | 913.8202 | GO, 631.5587 | OO, 603.5285 | 1.9 | |
| 42 | C22:1OO | 3 | 52 | 941.8532 | 941.8515 | C22:1O, 659.5901 | OO, 603.5278 | 1.8 | |
| 43 | C24:1LP | 3 | 52 | 941.8532 | 941.8515 | C24:1L, 685.6038 | LP, 575.4989 | C24:1P, 661.6040 | 1.8 |
| 44 | C22:1OG | 3 | 54 | 969.8845 | 969.8789 | C22:1O, 659.5889 | OG, 631.5591 | C22:1G, 687.6163 | 5.8 |
| 45 | C24:1OO | 3 | 54 | 969.8845 | 969.8789 | C24:1O, 687.6163 | OO, 603.5286 | 5.8 | |
| 46 | C24:1LS | 3 | 54 | 969.8845 | 969.8789 | C24:1L, 685.6038 | LS, 603.5286 | C24:1S, 689.6339 | 5.8 |
| 47 | LgLO | 3 | 54 | 969.8845 | 969.8789 | LgL, 687.6163 | OL, 601.5146 | LgO, 689.6339 | 5.8 |
| 48 | C22:1OC22:1 | 3 | 56 | 997.9158 | nd | C22:1O, 659.5894 | C22:1C22:1, 715.6480 | - | |
| 49 | C24:1OG | 3 | 56 | 997.9158 | nd | C24:1O, 687.6221 | OG, 631.5586 | C24:1G, 715.6480 | - |
| 50 | LLP | 4 | 44 | 855.7436 | 855.7401 | LP, 575.5005 | LL, 599.4967 | 4.1 | |
| 51 | γLnOP | 4 | 44 | 855.7436 | 855.7401 | OγLn, 599.4967 | PγLn, 573.4853 | PO, 577.5146 | 4.1 |
| 52 | OLO | 4 | 46 | 883.7749 | 883.7694 | OO, 603.5799 | OL, 601.5135 | 6.2 | |
| 53 | SLL | 4 | 46 | 883.7749 | 883.7694 | LL, 599.4969 | SL, 603.5799 | 6.2 | |
| 54 | GγLnP | 4 | 46 | 883.7749 | 883.7694 | GγLn, 627.5231 | PγLn, 573.4848 | GP, 605.5386 | 6.2 |
| 55 | SOγLn | 4 | 46 | 883.7749 | 883.7694 | OγLn, 599.4969 | SγLn, 601.5135 | SO, 605.5386 | 6.2 |
| 56 | GLO | 4 | 48 | 911.8062 | 911.7993 | GO, 631.5567 | OL, 601.5146 | GL, 629.5449 | 7.6 |
| 57 | C22:1γLnP | 4 | 48 | 911.8062 | 911.7993 | C22:1γLn, 655.5573 | γLnP, 573.4834 | C22:1P, 633.5735 | 7.6 |
| 58 | ALL | 4 | 48 | 911.8062 | 911.7993 | LL, 599.5042 | AL, 631.5567 | 7.6 | |
| 59 | GγLnS | 4 | 48 | 911.8062 | 911.7993 | GγLn, 627,5336 | SγLn, 601.5146 | GS, 633.5735 | 7.6 |
| 60 | C22:1LO | 4 | 50 | 939.8375 | 939.8345 | C22:1L, 657.5740 | LO, 601.5142 | C22:1O, 659.5886 | 3.2 |
| 61 | GLG | 4 | 50 | 939.8375 | 939.8345 | GL, 629.5450 | GG, 659.5886 | 3.2 | |
| 62 | BLL | 4 | 50 | 939.8375 | 939.8345 | BL, 659.5886 | LL, 599.5040 | 3.2 | |
| 63 | C24:1γLnP | 4 | 50 | 939.8375 | 939.8345 | C24:1γLn, 683.5854 | PγLn, 573.4833 | C24:1P, 661.6029 | 3.2 |
| 64 | C24:1LO | 4 | 52 | 967.8688 | 967.8639 | C24:1L, 685.6038 | LO, 601.5137 | C24:1O, 687.6177 | 5.1 |
| 65 | LgLL | 4 | 52 | 967.8688 | 967.8639 | LgL, 687.6177 | LL, 599.4961 | 5.1 | |
| 66 | C24:1γLnS | 4 | 52 | 967.8688 | 967.8639 | C24:1γLn, 683.5891 | SγLn, 601.5137 | C24:1S, 689.6338 | 5.1 |
| 67 | C22:1LC22:1 | 4 | 54 | 995.9001 | 995.8919 | C22:1L, 657.5935 | C22:1C22:1, 715.6518 | 8.2 | |
| 68 | C24:1LG | 4 | 54 | 995.9001 | 995.8919 | C24:1G, 715,6449 | GL, 629.5437 | C24:1L, 685.6041 | 8.2 |
| 69 | C24:1LC22:1 | 4 | 56 | 1023.9314 | nd | C24:1C22:1, 743.7023 | C22:1L, 657.5960 | C24:1L, 685.6038 | - |
| 70 | γLnLP | 5 | 42 | 853.7280 | 853.7251 | PγLn, 573.4852 | LP, 575.4991 | LγLn, 597.4786 | 3.4 |
| 71 | OLL | 5 | 44 | 881.7593 | 881.7557 | LL, 599.4979 | LO, 601.5102 | 4.1 | |
| 72 | OOγLn | 5 | 44 | 881.7593 | 881.7557 | OO, 603.5791 | OγLn, 599.4979 | 4.1 | |
| 73 | SLγLn | 5 | 44 | 881.7593 | 881.7557 | SL, 603.5791 | LγLn, 597.4829 | SγLn, 601.5102 | 4.1 |
| 74 | GLL | 5 | 46 | 909.7906 | 909.7893 | LL, 599.4985 | GL, 629.5434 | 1.4 | |
| 75 | GOγLn | 5 | 46 | 909.7906 | 909.7893 | GO, 631.5538 | OγLn, 599.4985 | GγLn, 627.5617 | 1.4 |
| 76 | ALγLn | 5 | 46 | 909.7906 | 909.7893 | AL, 631.5538 | LγLn, 597.4820 | ALn, 629.5434 | 1.4 |
| 77 | C22:1LL | 5 | 48 | 937.8219 | 937.8167 | LL, 599.4983 | C22:1L, 657.5733 | 5.5 | |
| 78 | BLγLn | 5 | 48 | 937.8219 | 937.8167 | BL, 659.5866 | LγLn, 597.4889 | BLn, 657.5733 | 5.5 |
| 79 | C24:1LL | 5 | 50 | 965.8532 | 965.8522 | C24:1L, 685.6172 | LL, 599.4981 | 1.0 | |
| 80 | C24:1OγLn | 5 | 50 | 965.8532 | 965.8522 | C24:1O, 687.6172 | OγLn, 599.4981 | C24:1γLn, 683.5902 | 1.0 |
| 81 | C22:1γLnC22:1 | 5 | 52 | 993.8845 | 993.8809 | C22:1γLn, 655.5937 | C22:1C22:1, 715,6605 | 3.6 | |
| 82 | γLnγLnP | 6 | 40 | 851.7123 | 851.7112 | γLnγLn, 595.4679 | PγLn, 573.4847 | 1.3 | |
| 83 | LLL | 6 | 42 | 879.7436 | 879.7394 | LL, 599.4968 | 4.8 | ||
| 84 | OLγLn | 6 | 42 | 879.7436 | 879.7394 | OL, 601.5088 | LγLn, 597.4845 | OγLn, 599.4968 | 4.8 |
| 85 | SγLnγLn | 6 | 42 | 879.7436 | 879.7394 | γLnγLn, 595.4684 | SγLn, 601.5088 | 4.8 | |
| 86 | GLγLn | 6 | 44 | 907.7749 | 907.7733 | GL, 629.5408 | LγLn, 597.4845 | GγLn, 627.5283 | 1.8 |
| 87 | C22:1LγLn | 6 | 46 | 935.8062 | 935.8065 | C22:1L, 657.5728 | LγLn, 597.4825 | C22:1γLn, 655.5612 | 0.3 |
| 88 | C24:1LγLn | 6 | 48 | 963.8375 | 963.8337 | C24:1L, 685.6049 | LγLn, 597.4802 | C24:1γLn, 683.5907 | 3.9 |
| 89 | γLnγLnPo | 7 | 38 | 849.6967 | 849.6957 | γLnγLn, 595.4453 | PoγLn, 571.4680 | 1.2 | |
| 90 | LLγLn | 7 | 40 | 877.7280 | 877.7222 | γLnL, 597.4834 | LL, 599.4971 | 6.6 | |
| 91 | γLnOγLn | 7 | 40 | 877.7280 | 877.7222 | OγLn, 599.4971 | γLnγLn, 595.4707 | 6.6 | |
| 92 | GγLnγLn | 7 | 42 | 905.7593 | 905.7592 | GγLn, 627.5271 | γLnγLn, 595.4721 | 0.1 | |
| 93 | γLnLγLn | 8 | 38 | 875.7123 | 875.7076 | LγLn, 597.4819 | γLnγLn, 595.4669 | 5.4 | |
| 94 | γLnγLnγLn | 9 | 36 | 873.6967 | 873.6966 | γLnγLn, 595.4670 | 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arena, P.; Sciarrone, D.; Dugo, P.; Donato, P.; Mondello, L. Pattern-Type Separation of Triacylglycerols by Silver Thiolate×Non-Aqueous Reversed Phase Comprehensive Liquid Chromatography. Separations 2021, 8, 88. https://doi.org/10.3390/separations8060088
Arena P, Sciarrone D, Dugo P, Donato P, Mondello L. Pattern-Type Separation of Triacylglycerols by Silver Thiolate×Non-Aqueous Reversed Phase Comprehensive Liquid Chromatography. Separations. 2021; 8(6):88. https://doi.org/10.3390/separations8060088
Chicago/Turabian StyleArena, Paola, Danilo Sciarrone, Paola Dugo, Paola Donato, and Luigi Mondello. 2021. "Pattern-Type Separation of Triacylglycerols by Silver Thiolate×Non-Aqueous Reversed Phase Comprehensive Liquid Chromatography" Separations 8, no. 6: 88. https://doi.org/10.3390/separations8060088