
Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses


 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microextraction Techniques
Solid-Phase Extraction (SPE)
2.2. Solid-Phase Microextraction
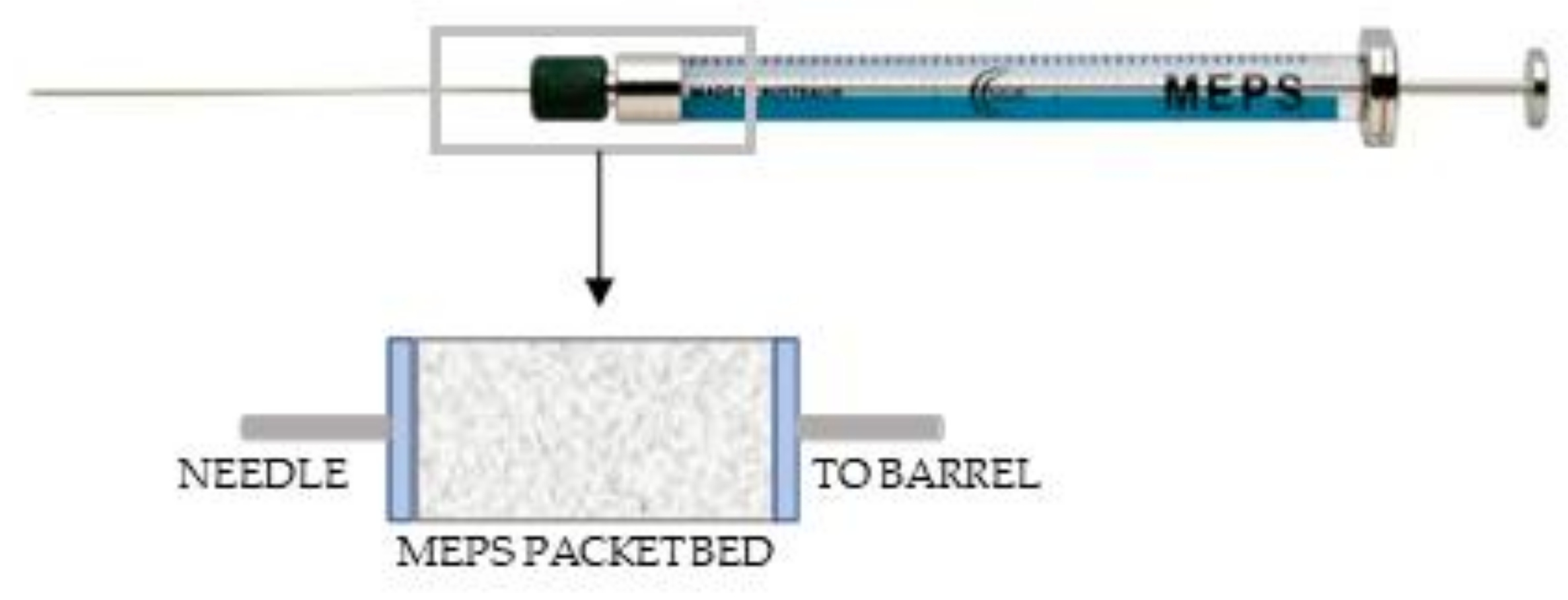
2.3. Microextraction by Packed Sorbent (MEPS)
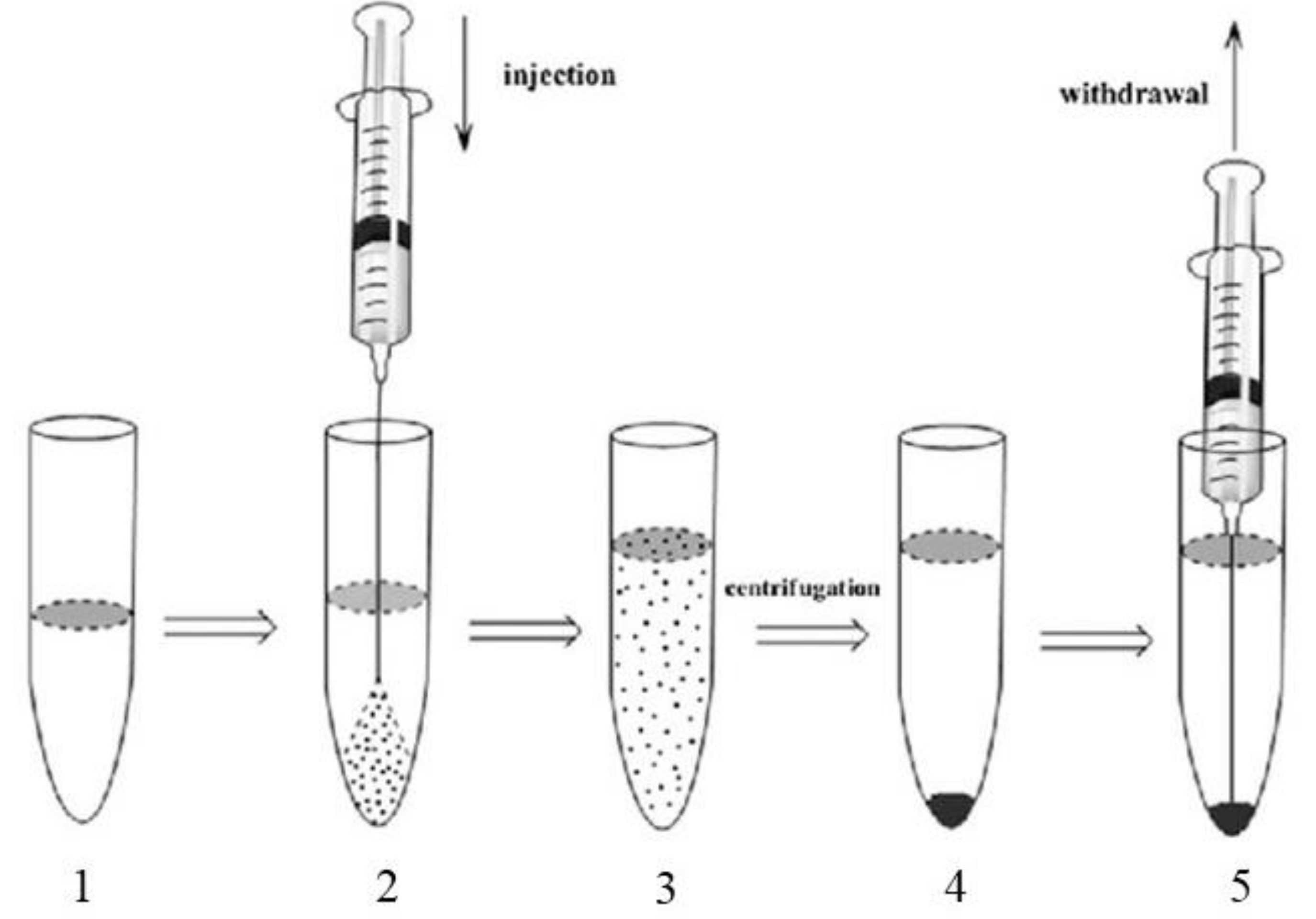
2.4. Liquid-Liquid Extraction (LLE)
2.5. Liquid-Phase Microextraction (LPME)
2.6. Other Techniques
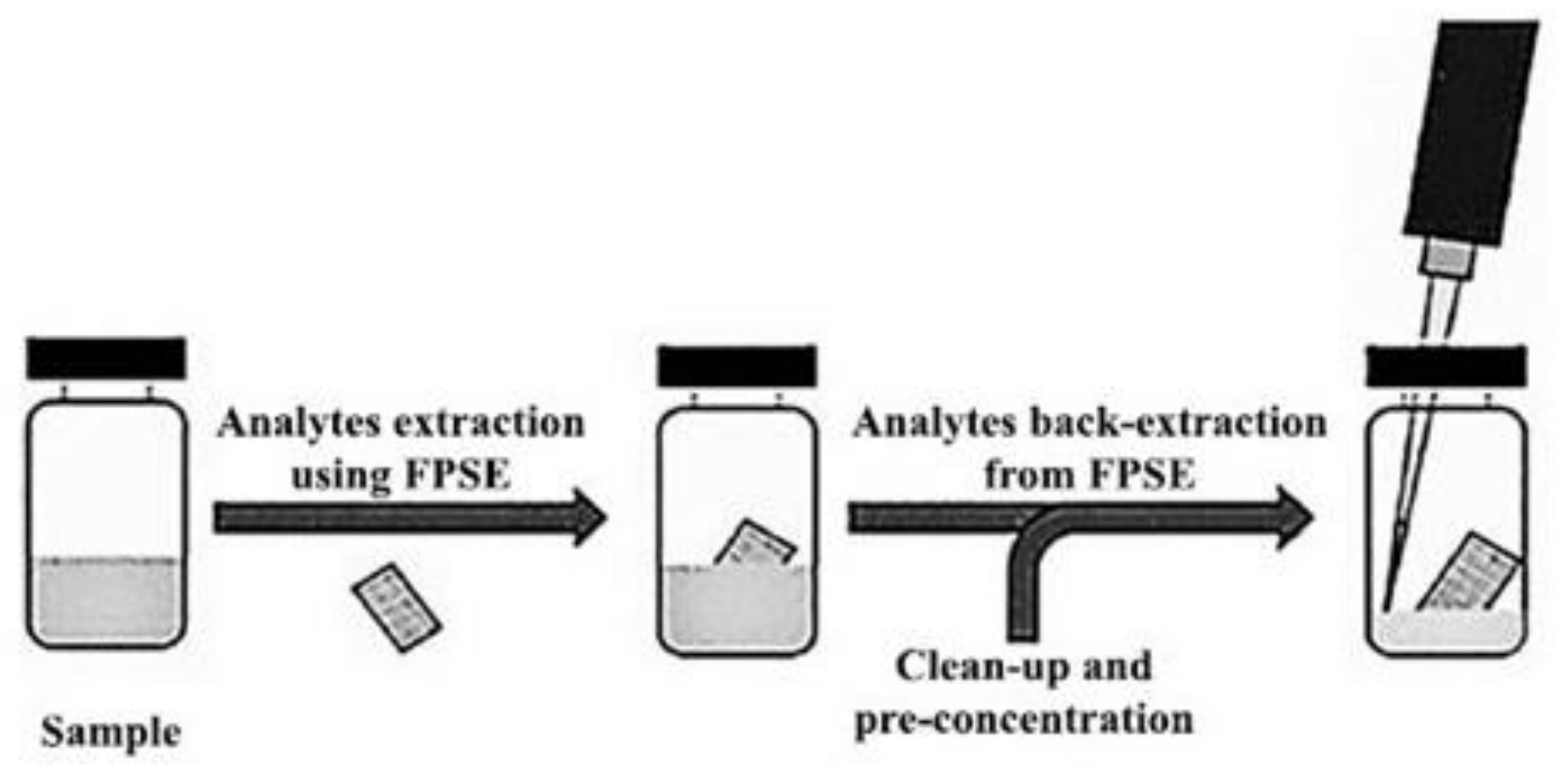
2.7. Fabric Phase Sorptive Extraction (FPSE) as an Innovative Procedure in the Biological Field
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pourshamsi, T.; Amri, F.; Abniki, M. A comprehensive review on application of the syringe in liquidand solid phase microextraction methods. J. Iran. Chem. Soc. 2021, 18, 245–264. [Google Scholar] [CrossRef]
- Hansen, F.A.; Pedersen-Bjergaard, S. Emerging Extraction Strategies in Analytical Chemistry. Anal. Chem. 2020, 92, 2–15. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Concheiro-Guisan, M. Microextraction sample preparation techniques in forensic analytical toxicology. Biomed. Chromatogr. 2019, 33, e4444. [Google Scholar] [CrossRef] [Green Version]
- Manousi, N.; Samanidou, V. Green sample preparation of alternative biosamples in forensic toxicology. Sustain. Chem. Pharm. 2021, 20, e100388. [Google Scholar] [CrossRef]
- D’Ovidio, C.; Rosato, E.; Bonelli, M.; Carnevale, A.; Marsella, L.T. A particular case of accidental asphyxiation. Med. Sci. Law. 2018, 58, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ovidio, C.; Bonelli, M.; Rosato, E.; Savini, F.; Carnevale, A. Evaluation of urinary catecholamines to reconstruct the individual death process after the catastrophe of Rigopiano (Italy). J. Forensic Leg. Med. 2020, 70, 101908. [Google Scholar] [CrossRef] [PubMed]
- D’Ovidio, C.; Rosato, E.; Carnevale, A. An unusual case of murder-suicide: The importance of studying knots. J. Forensic Leg. Med. 2017, 45, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Majda, A.; Mrochem, K.; Wietecha-Posłuszny, R.; Zapotoczny, S.; Zawadzki, M. Fast and efficient analyses of the post-mortem human blood and bone marrow using DI-SPME/LC-TOF-MS method for forensic medicine purposes. Talanta 2020, 209, e120533. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Gonçalves, O.C.; Oliveira, M.N.; Neng, N.R.; Nogueira, J.M.F. Application of Microextraction-Based Techniques for Screening-Controlled Drugs in Forensic Context—A Review. Molecules 2021, 26, 2168. [Google Scholar] [CrossRef] [PubMed]
- Seyed, M.S.A.; Mohammad, J.K.; Zahra, M.; Maryam, A. Comparison of the Modified QuEChERS Method and the Conventional Method of Extraction in Forensic Medicine to Detect Methadone in Post-Mortem Urine by GC-MS. Asia Pac. J. Med. Toxicol. 2017, 6, 79–85. [Google Scholar] [CrossRef]
- Locatelli, M.; Tartaglia, A.; Piccolantonio, S.; Di Iorio, L.A.; Sperandio, E.; Ulusoy, H.I.; Furton, K.G.; Kabir, A. Innovative configurations of sample preparation Techniques Applied in bioanalytical chemistry: A review. Curr. Anal. Chem. 2019, 15, 731–744. [Google Scholar] [CrossRef]
- Sofalvi, S.; Schueler, H.E.; Lavins, E.S.; Kaspar, C.K.; Brooker, I.T.; Mazzola, C.D.; Dolinak, D.; Gilson, T.P.; Perch, S. An LC-MS/MS Method for the Analysis of Carfentanil, 3-Methylfentanyl, 2-Furanyl Fentanyl, Acetyl Fentanyl, Fentanyl and Norfentanyl in Postmortem and Impaired-Driving Cases. J. Anal. Toxicol. 2017, 41, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Kahl, J.H.; Gonyea, J.; Humphrey, S.M.; Hime, G.W.; Boland, D.M. Quantitative Analysis of Fentanyl and Six Fentanyl Analogs in Postmortem Specimens by UHPLC-MS/MS. J. Anal. Toxicol. 2018, 42, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.F.; Papsun, D.M.; Logan, B.K. Analysis of Fentanyl and 18 Novel Fentanyl Analogs and Metabolites by LC-MS/MS, and report of Fatalities Associated with Methoxyacetylfentanyl and Cyclopropylfentanyl. J. Anal. Toxicol. 2018, 42, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Palmquist, K.B.; Swortwood, M.J. Data-independent screening method for 14 fentanyl analogs in whole blood and oral fluid using LC-QTOF-MS. Forensic Sci. Int. 2019, 297, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Strayer, K.E.; Antonides, H.M.; Juhascik, M.P.; Daniulaityte, R.; Sizemore, I.E. LC-MS/MS-Based Method for the Multiplex Detection of 24 Fentanyl Analogues and Metabolites in Whole Blood at Sub ng mL−1 Concentrations. ACS Omega 2018, 3, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Truver, M.T.; Swortwood, M.J. Quantitative Analysis of Novel Synthetic Opioids, Morphine and Buprenorphine in Oral Fluid by LC-MS/MS. J. Anal. Toxicol. 2018, 42, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Truver, M.T.; Swortwood, M.J. Quantification of U-47700 and its metabolites in plasma by LC-MS/MS. J. Chromatogr. B 2019, 1112, 41–47. [Google Scholar] [CrossRef]
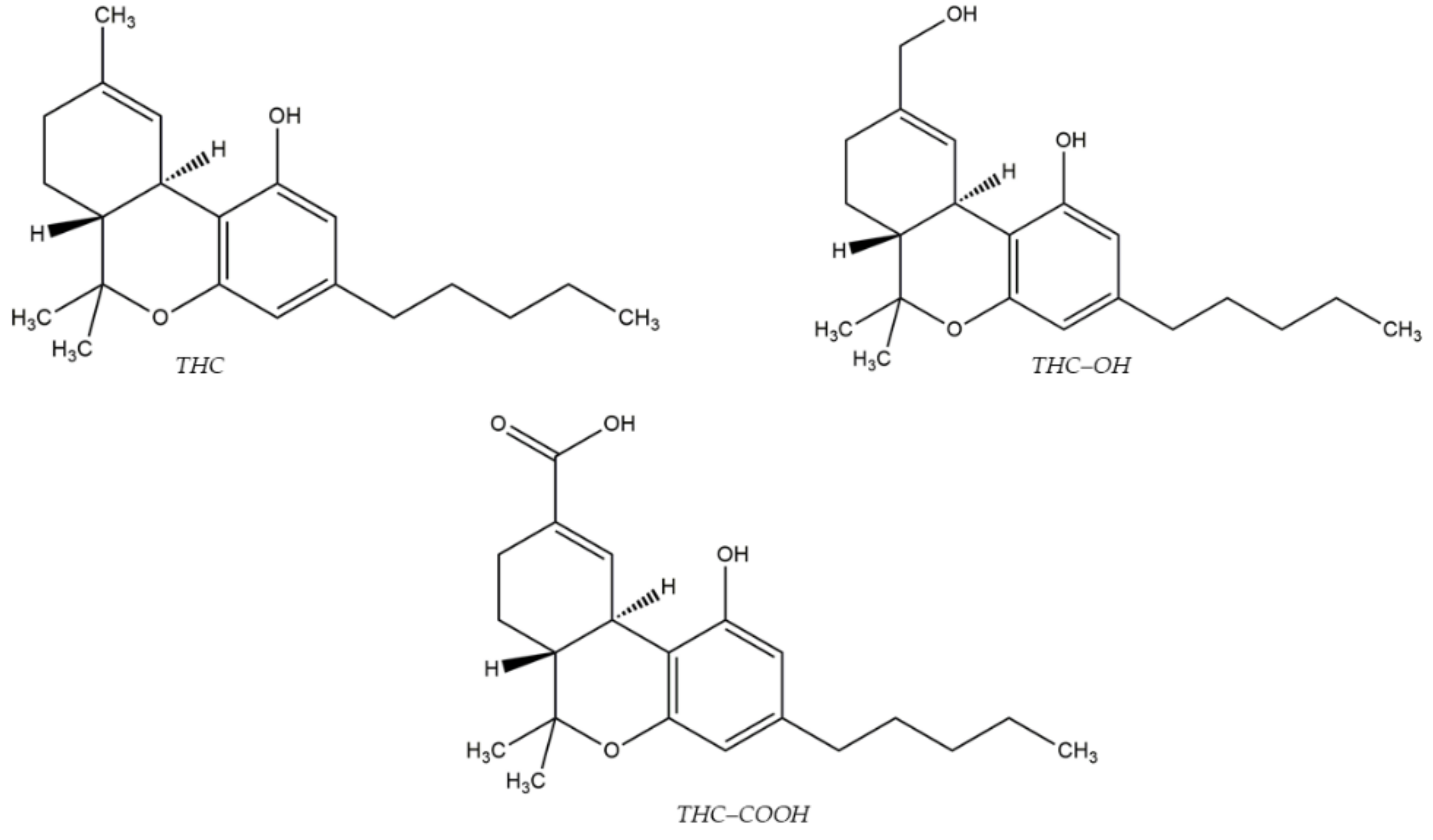
- Al-Asmari, A.I. Method for Postmortem Quantification of Δ9-Tetrahydrocannabinol and Metabolites Using LC-MS/MS. J. Anal. Toxicol. 2019, 43, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Al-Asmari, A.I. Method for the identification and quantification of sixty drugs and their metabolites in postmortem whole blood using liquid chromatography tandem mass spectrometry. Forensic Sci. Int. 2020, 309, e110193. [Google Scholar] [CrossRef]
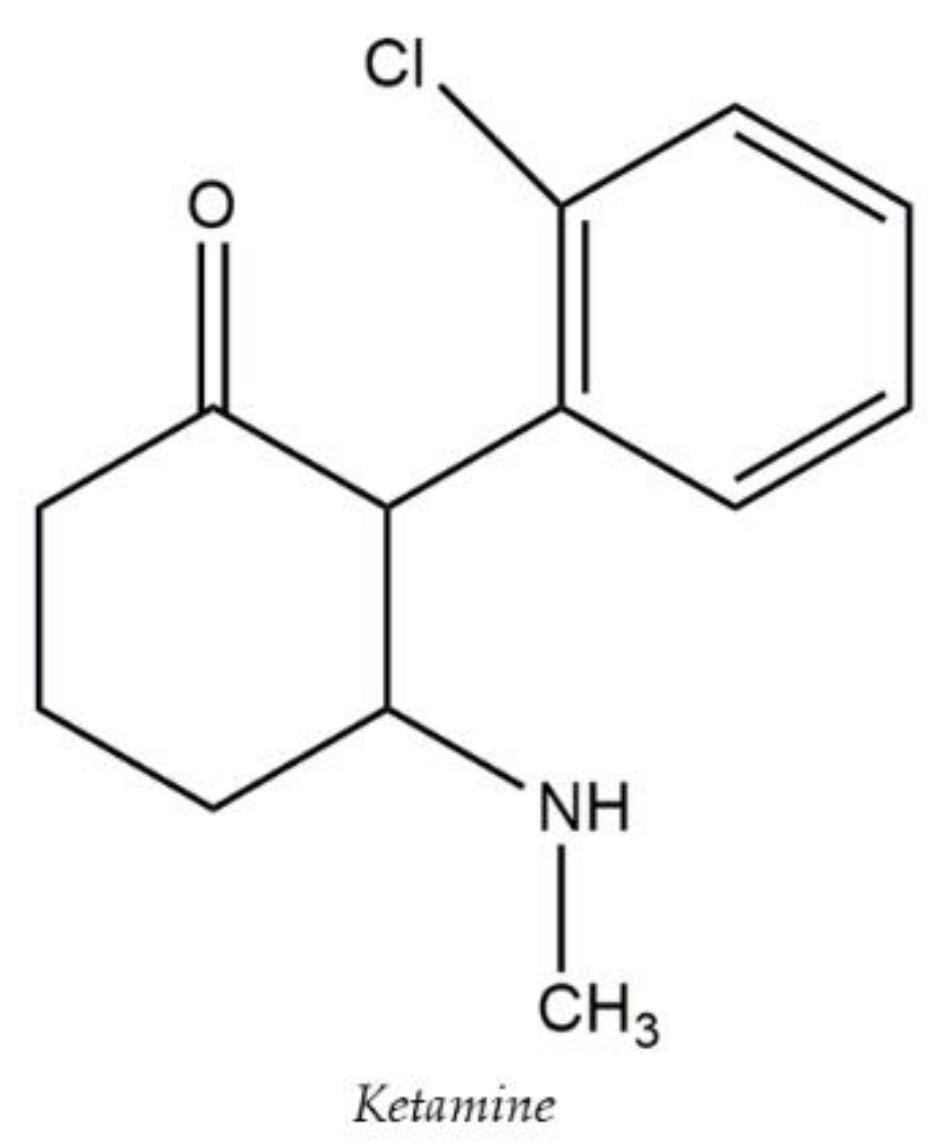
- Kurzweil, L.; Danyeli, L.; Şen, Z.D.; Fejtova, A.; Walter, M.; Gensberger-Reigl, S. Targeted mass spectrometry of ketamine and its metabolites cis-6-hydroxynorketamine and norketamine in human blood serum. J. Chromatogr. B 2020, 1152, e122214. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, D.; Zhao, M.; Li, J.; Yang, Y.; Li, M.; Gao, J.; Jiang, Y. A fast and simple approach for the quantification of fiveanti-hypersensitivity drugs in saliva and urine by portable ionmobility spectrometry based on magnetic graphene oxide dispersivesolid phase extraction. J. Pharm. Biomed. 2020, 189, e113414. [Google Scholar] [CrossRef]
- Giebułtowicz, J.; Sobiech, M.; Ruzycka, M.; Lulinski, P. Theoretical and experimental approach to hydrophilic interactiondispersive solid-phase extraction of 2-aminothiazoline-4-carboxylicacid from human post-mortem blood. J. Chromatogr. A 2019, 1587, 61–72. [Google Scholar] [CrossRef]
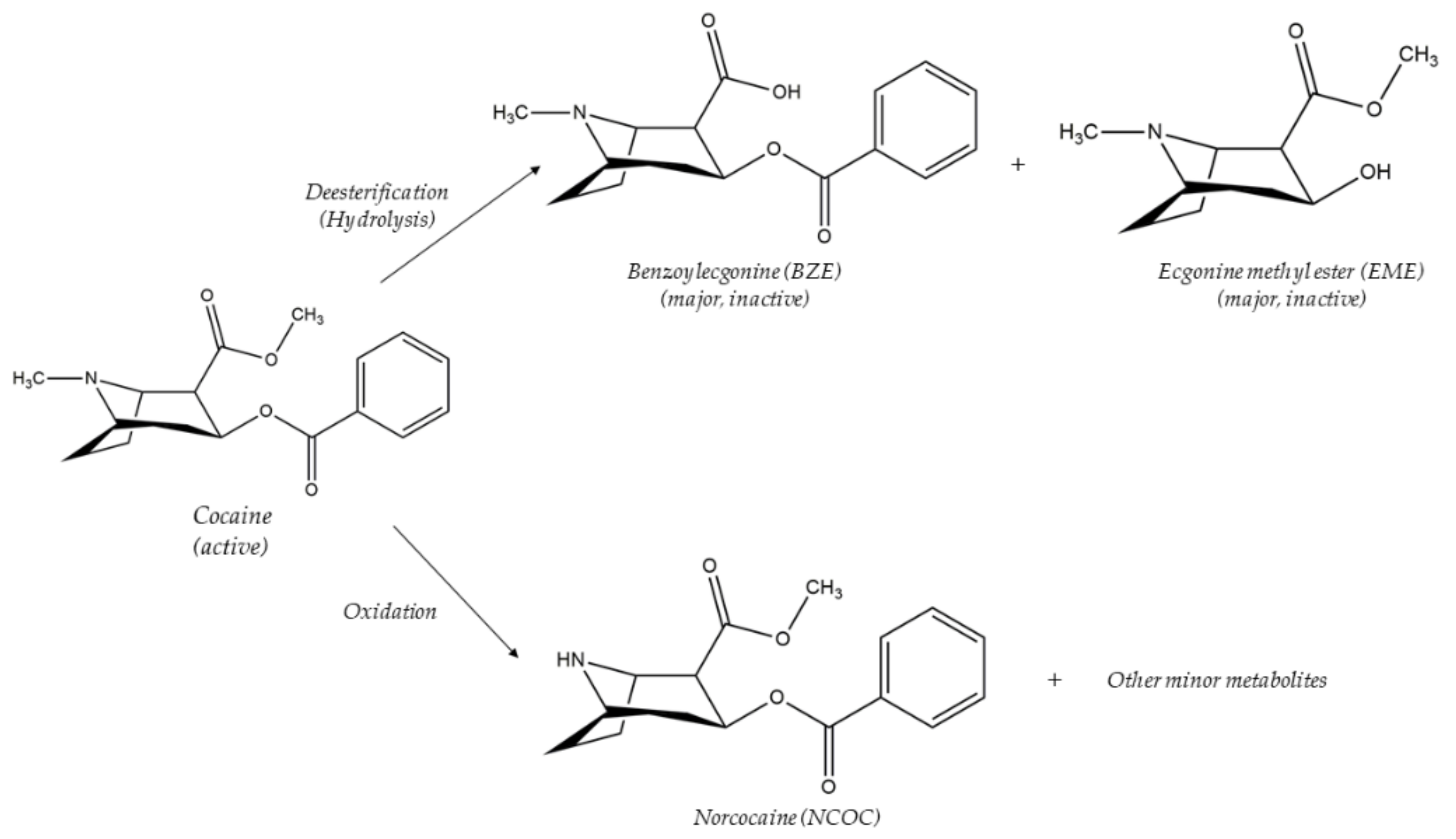
- Feltraco Lizot, L.; Cezimbra da Silva, A.C.; Bastiani, M.F.; Maurer, T.F.; Hahn, R.Z.; Perassolo, M.S.; Antunes, M.V.; Linden, R. Simultaneous Determination of Cocaine and Metabolites in Human Plasma Using Solid Phase Micro-Extraction Fiber Tips C18 and UPLC-MS/MS. J. Anal. Toxicol. 2020, 44, 49–56. [Google Scholar] [CrossRef]
- Nakhodchi, S.; Alizadeh, N. Rapid simultaneous determination of ketamine and midazolam in biological samples using ion mobility spectrometry combined by headspace solid-phase microextraction. J. Chromatogr. A 2021, 1658, e462609. [Google Scholar] [CrossRef] [PubMed]
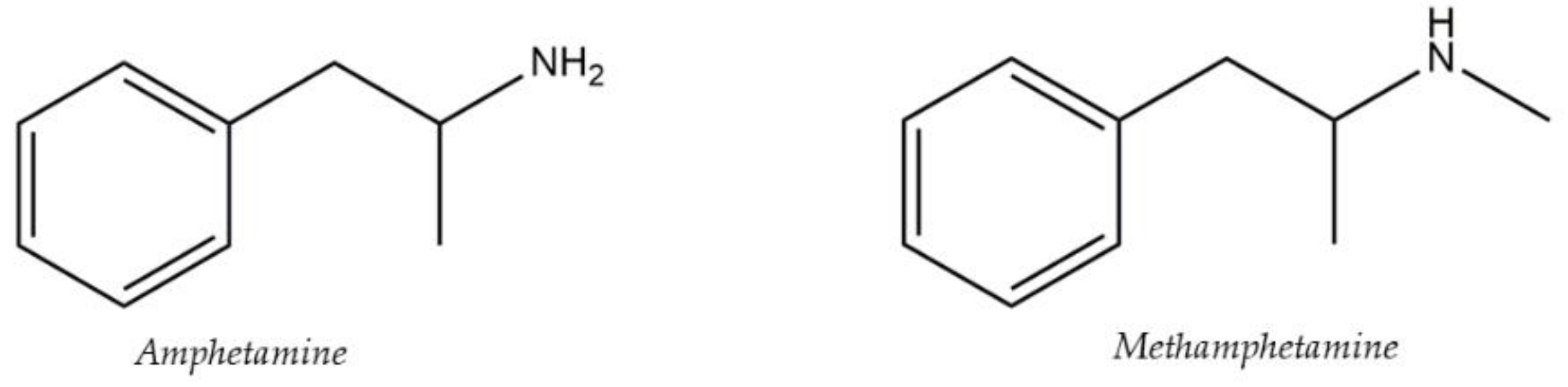
- Song, A.; Wang, J.; Lu, G.; Jia, Z.; Yang, J.; Shi, E. Oxidized multiwalled carbon nanotubes coated fibers for headspace solid-phase microextraction of amphetamine-type stimulants in human urine. Forensic Sci. Int. 2018, 290, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.; González, M.; Regenjo, M.; Ares, A.M.; Fernández, A.M.; Lorenzo, R.A.; Carro, A.M. Analysis of drugs of abuse in human plasma using microextraction bypacked sorbents and ultra-high-performance liquid chromatography. J. Chromatogr. A 2017, 1485, 8–19. [Google Scholar] [CrossRef]
- Prata, M.; Ribeiro, A.; Figueirinha, D.; Rosado, T.; Oppolzer, D.; Restolho, J.; Araújo Suzel Costa, A.R.T.S.; Barroso, M.; Gallardo, E. Determination of opiates in whole blood using microextraction bypacked sorbent and gas chromatography-tandem mass spectrometry. J. Chromatogr. A 2019, 1602, 1–10. [Google Scholar] [CrossRef]
- Sorribes-Soriano, A.; Monedero, A.; Esteve-Turrillas, F.A.; Armenta, S. Determination of the new psychoactive substance dichloropane insaliva by microextraction by packed sorbent—Ion mobility spectrometry. J. Chromatogr. A 2019, 1603, 61–66. [Google Scholar] [CrossRef]
- Vincenti, F.; Montesano, C.; Pirau, S.; Gregori, A.; Di Rosa, F.; Curini, R.; Sergi, M. Simultaneous Quantification of 25 Fentanyl Derivatives and Metabolites in Oral Fluid by Means of Microextraction on Packed Sorbent and LC-HRMS/MS Analysis. Molecules 2021, 26, 5870. [Google Scholar] [CrossRef] [PubMed]
- Vejar-Vivar, C.; Bustamante, L.; Lucena, R.; Ortega, C.; Valenzuela, M.; Mardones, C. Direct coupling of MEPS to ESI-QqTOF-MS for the simultaneous analysis of tricyclic antidepressants and benzodiazepines in postmortem blood. Microchem. J. 2021, 171, e106797. [Google Scholar] [CrossRef]
- Da Cunha, K.F.; Rodrigues, L.C.; Huestis, M.A.; Costa, J.L. Miniaturized extraction method for analysis of synthetic opioids in urine by microextraction with packed sorbent and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2020, 1624, e461241. [Google Scholar] [CrossRef]
- Degreef, M.; van Nuijs, A.L.N.; Maudens, K.E. Validation of a simple, fast liquid chromatography-tandem mass spectrometry method for the simultaneous quantification of 40 antidepressant drugs or their metabolites in plasma. Clin. Chim. Acta 2018, 485, 243–257. [Google Scholar] [CrossRef]
- Zawadzki, M.; Kowalski, G.; Konowałek, A.C.; Siczek, M.; Sobieszczanska, M.; Leppert, W.; Wieczorowska-Tobis, K.; Szpot, P. Rapid Determination of Sufentanil in Human Plasma by UHPLC-QqQ-MS/MS. J. Anal. Toxicol. 2021, 45, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Bombana, H.S.; dos Santos, M.F.; Munoz, D.R.; Leyton, V. Hollow-fibre liquid-phase microextraction and gas chromatography-mass spectrometric determination of amphetamines in whole blood. J. Chromatogr. B 2020, 1139, e121973. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Montesano, C.; Cellucci, L.; Gregori, A.; Fanti, F.; Compagnone, D.; Curini, R.; Sergi, M. Combination of pressurized liquid extraction with dispersive liquid liquid micro extraction for the determination of sixty drugs of abuse in hair. J. Chromatogr. A 2019, 1605, e360348. [Google Scholar] [CrossRef] [PubMed]
- Mercieca, G.; Odoardi, S.; Cassar, M.; Rossi, S.S. Rapid and simple procedure for the determination of cathinones, amphetamine-like stimulants and other new psychoactive substancesin blood and urine by GC-MS. J. Pharm. Biomed. 2018, 149, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Temerdashev, A.; Dmitrieva, E.; Azaryan, A.; Gashimova, E. A novel approach to the quantification of urinary arylpropionamide-derived SARMs by UHPLC-MS/MS. Biomed. Chromatogr. 2020, 34, e4700. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, M.; Dubrulle, L.; Dehaen, W.; Tytgat, J.; Cuypers, E. Fast and easy extraction of antidepressants from whole blood using ionic liquids as extraction solvent. Talanta 2018, 180, 292–299. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, M.; Dehaen, W.; Tytgat, J.; Cuypers, E. Ionic Liquid-Based Liquid-Liquid Microextraction for Benzodiazepine Analysis in Postmortem Blood Samples. J. Forensic Sci 2018, 63, 1875–18709. [Google Scholar] [CrossRef] [PubMed]
- Vårdal, L.; Wong, G.; Øiestad, A.M.L.; Pedersen-Bjergaard, S.; Gjelstad, A.; Øiestad, E.L. Rapid determination of designer benzodiazepines, benzodiazepines, and Z-hypnotics in whole blood using parallel artificial liquid membrane extraction and UHPLC-MS/MS. Anal. Bioanal. Chem. 2018, 410, 4967–4978. [Google Scholar] [CrossRef]
- Ask, K.S.; Lid, M.; Øiestad, E.L.; Pedersen-Bjergaard, S.; Gjelstad, A. Liquid-phase microextraction in 96-well plates-calibration and accurate quantification of pharmaceuticals in human plasma samples. J. Chromatogr. A 2019, 1602, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Orfanidis, A.; Gika, H.G.; Theodoridis, G.; Mastrogianni, O.; Raikos, N. A UHPLC MS-MS Method for the Determination of 84 Drugs of Abuse and Pharmaceuticals in Blood. J. Anal. Toxicol. 2021, 45, 28–43. [Google Scholar] [CrossRef]
- De Boeck, M.; Missotten, S.; Dehaen, W.; Tytgat, J.; Cuypers, E. Development and validation of a fast ionic liquid-based dispersive liquid-liquid microextraction procedure combined with LC-MS/MS analysis for the quantification of benzodiazepines and benzodiazepine-like hypnotics in whole blood. Forensic Sci. Int. 2017, 274, 44–54. [Google Scholar] [CrossRef]
- Caspar, A.T.; Kollas, A.B.; Maurer, H.H.; Meyer, M.R. Development of a quantitative approach in blood plasma for low-dosed hallucinogens and opioids using LC-high resolution mass spectrometry. Talanta. 2018, 176, 635–645. [Google Scholar] [CrossRef]
- Rocchi, R.; Simeoni, M.C.; Montesano, C.; Vannutelli, G.; Curini, R.; Sergi, M.; Compagnone, D. Analysis of new psychoactive substances in oral fluids by means of microextraction by packed sorbent followed by ultra-high performance liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2018, 10, 865–873. [Google Scholar] [CrossRef]
- Shoff, E.N.; Zaney, M.E.; Kahl, J.H.; Hime, G.W.; Boland, D.M. Qualitative Identification of Fentanyl Analogs and Other Opioids in Postmortem Cases by UHPLC-Ion Trap-MSn. J. Anal. Toxicol. 2017, 41, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, J.; Zhang, X.; Qiu, M.; Huang, Z.; Rao, Y. Ultrasound-assisted dispersive liquid-liquid microextraction for thedetermination of seven recreational drugs in human whole blood using gas chromatography-mass spectrometry. J. Chromatogr. B 2017, 1046, 177–184. [Google Scholar] [CrossRef]
- Rodrigues, T.B.; Morais, D.R.; Gianvecchio, V.A.P.; Aquino, E.M.; Cunha, R.L.; Huestis, M.A.; Costa, J.L. Development and Validation of a Method for Quantification of 28 Psychotropic Drugs in Postmortem Blood Samples by Modified Micro-QuEChERS and LC-MS-MS. J. Anal. Toxicol. 2021, 45, 644–656. [Google Scholar] [CrossRef]
- Scheid, C.; Eller, S.; Oenning, A.L.; Carasek, E.; Merib, J.; De Oliveira, T.F. Application of Homogeneous Liquid-Liquid Microextraction with Switchable Hydrophilicity Solvents to the Determination of MDMA, MDA and NBOMes in Postmortem Blood Samples. J. Anal. Toxicol. 2021, bkab100, IN PRESS. [Google Scholar] [CrossRef] [PubMed]
- Pouliopoulos, A.; Tsakelidou, E.; Krokos, A.; Gika, H.G.; Theodoridis, G.; Raikos, N. Quantification of 15 Psychotropic Drugs in Serum and Postmortem Blood Samples after a Modified Mini-QuEChERS by UHPLC-MS-MS. J. Anal. Toxicol. 2018, 42, 337–345. [Google Scholar] [CrossRef]
- Pires da Silva, C.; Dal Piaz, L.P.P.; Gerbase, F.E.; Vendramini Müller, V.; Lima Feltraco Lizot, L.; Venzon Antunes, M.; Linden, R. Simple extraction of toxicologically relevant psychotropic compounds and metabolites from whole blood using mini-QuEChERS followed by UPLC-MS/MS analysis. Biomed. Chromatogr. 2021, 35, e5142. [Google Scholar] [CrossRef]
- Kusano, M.; Sakamoto, Y.; Natori, Y.; Miyagawa, H.; Tsuchihashi, H.; Ishii, A.; Zaitsu, K. Development of “Quick-DB forensic”: A total workflow from QuEChERS-dSPE method to GC-MS/MS quantification of forensically relevant drugs and pesticides in whole blood. Forensic Sci. Int. 2019, 300, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Campêlo, J.M.; Rodrigues, T.B.; Costa, J.L.; Santos, J.M. Optimization of QuEChERS extraction for detection and quantification of 20 antidepressants in postmortem blood samples by LC-MS/MS. Forensic Sci. Int. 2021, 319, 110660. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Petrikovics, I.; Yu, J. Performance comparison between solid phase extraction and magnetic carbon nanotubes facilitated dispersive-micro solid phase extractions (Mag-CNTs/d-μSPE) of a cyanide metabolite in biological samples using GC-MS. J. Anal. Sci Technol. 2021, 12, 42. [Google Scholar] [CrossRef]
- Accioni, F.; García-Gómez, D.; Girela, E.; Rubio, S. SUPRAS extraction approach for matrix-independent determination of amphetamine-type stimulants by LC-MS/MS. Talanta 2018, 182, 574–582. [Google Scholar] [CrossRef]
- Gallo, V.; Tomai, P.; Di Lisio, V.; Dal Bosco, C.; D’Angelo, P.; Fanali, C.; D’Orazio, G.; Silvestro, I.; Picó, Y.; Gentili, A. Application of a Low Transition Temperature Mixture for the Dispersive Liquid-Liquid Microextraction of Illicit Drugs from Urine Samples. Molecules 2021, 26, 5222. [Google Scholar] [CrossRef] [PubMed]
- Tomai, P.; Gentili, A.; Curini, R.; Gottardo, R.; Tagliaro, F.; Fanali, S. Dispersive liquid-liquid microextraction, an effective tool for the determination of synthetic cannabinoids in oral fluid by liquid chromatography tandem mass spectrometry. J. Pharm. Anal. 2021, 11, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Shishov, A.Y.; Chislov, M.V.; Nechaeva, D.V.; Moskvin, L.N.; Bulatov, A.V. A new approach for microextraction of non-steroidal anti-inflammatory drugs from human urine samples based on in-situ deep eutectic mixture formation. J. Mol. Liq. 2018, 272, 738–745. [Google Scholar] [CrossRef]
- Zamani, F.; Farajmand, B.; Yaftian, M.R. Corona discharge ion mobility spectrometry combined by homogenizer assisted dispersive liquid-phase microextraction; A rapid and sensitive method for quantification of nortriptyline. Microchem. J. 2020, 159, e105540. [Google Scholar] [CrossRef]
- Barati, E.; Alizadeh, N. Simultaneous determination of sertraline, imipramine and alprazolam in human plasma samples using headspace solid phase microextraction based on a nanostructured polypyrrole fiber coupled to ion mobility spectrometry. Anal. Methods 2020, 12, 930–937. [Google Scholar] [CrossRef]
- Khelfi, A.; Azzouz, M.; Abtroun, R.; Reggabi, M.; Alamir, B. Determination of Chlorpromazine, Haloperidol, Levomepromazine, Olanzapine, Risperidone, and Sulpiride in Human Plasma by Liquid Chromatography/Tandem Mass Spectrometry (LC-MS/MS). Int. J. Anal. Chem. 2018, 2018, e5807218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degreef, M.; Berry, E.M.; Maudens, K.E.K.; van Nuijs, A.L.N. Multi-analyte LC-MS/MS quantification of 38 antipsychotics and metabolites in plasma: Method validation & application to routine analyses. J. Chromatogr. B 2021, 1179, e122867. [Google Scholar] [CrossRef]
- Locatelli, M.; Tinari, N.; Grassadonia, A.; Tartaglia, A.; Macerola, D.; Piccolantonio, S.; Sperandio, E.; D’Ovidio, C.; Carradori, S.; Ulusoy, H.I.; et al. FPSE-HPLC-DAD method for the quantification of anticancer drugs in human whole blood, plasma, and urine. J. Chromatogr. B 2018, 1095, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Rochani, A.; Lam, E.; Tanjuakio, J.; Hirose, H.; Kraft, W.K.; Kaushal, G. Simultaneous quantitative LC-MS method of ketamine, midazolamand their metabolites (dehydronorketamine, norketamine and 1hydroxymidazolam) for its application in patients on extracorporealmembrane oxygenation (ECMO) therapy. J. Pharm. Biomed. 2020, 178, e112947. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Y.; Liu, X.; Fu, R.; Yang, L. Rapid and sensitive tapered-capillary microextraction combined to on-line sample stacking-capillary electrophoresis for extraction and quantification of two beta-blockers in human urine. Talanta 2018, 180, 90–97. [Google Scholar] [CrossRef]
- Nazdrajić, E.; Tascon, M.; Rickert, D.A.; Gómez-Ríos, G.A.; Kulasingam, V.; Pawliszyn, J.B. Rapid determination of tacrolimus and sirolimus in whole human blood by direct coupling of solid-phase microextraction to mass spectrometry via microfluidic open interface. Anal. Chim. Acta 2021, 1144, 53–60. [Google Scholar] [CrossRef]
- Locatelli, M.; Kabir, A.; Innosa, D.; Lopatriello, T.; Furton, K.G. A fabric phase sorptive extraction-High performance liquid chromatography-Photo diode array detection method for the determination of twelve azole antimicrobial drug residues in human plasma and urine. J. Chromatogr. B 2017, 1040, 192–198. [Google Scholar] [CrossRef]
- Hasan, M.; Hofstetter, R.; Fassauer, G.M.; Link, A.; Siegmund, W.; Oswald, S. Quantitative chiral and achiral determination of ketamine and itsmetabolites by LC-MS/MS in human serum, urine and fecal samples. J. Pharm. Biomed. Anal. 2017, 139, 87–97. [Google Scholar] [CrossRef]
- Jouyban, A.; Farajzadeh, M.A.; Mogaddam, M.R.A.; Khodadadeian, F.; Nemati, M.; Khoubnasabjafari, M. In-situ formation of a hydrophobic deep eutectic solvent based on alpha terpineol and its application in liquid-liquid microextraction of three β-blockers from plasma samples. Microchem. J. 2021, 170, e106687. [Google Scholar] [CrossRef]
- Šlampova, A.; Kubaň, P. Two-phase micro-electromembrane extraction with a floating drop free liquid membrane for the determination of basic drugs in complex samples. Talanta 2020, 206, 120255. [Google Scholar] [CrossRef]
- Kumar, R.; Gaurav; Heena; Malik, A.K.; Kabir, A.; Furton, K.G. Efficient analysis of selected estrogens using fabric phase sorptiveextraction and high-performance liquid chromatography-fluorescence detection. J. Chromatogr. A 2014, 1359, 16–25. [Google Scholar] [CrossRef]
- Guedes-Alonso, R.; Ciofi, L.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J.; del Bubba, M.; Kabir, A.; Furton, K.G. Determination of androgens and progestogens in environmental andbiological samples using fabric phase sorptive extraction coupled toultra-high performance liquid chromatography tandem massspectrometry. J. Chromatogr. A 2016, 1437, 116–126. [Google Scholar] [CrossRef]
- Samanidou, V.; Kaltzi, I.; Kabir, A.; Furton, K.G. Simplifying sample preparation using fabric phase sorptive extraction technique for the determination of benzodiazepines in blood serum by high-performance liquid chromatography. Biomed. Chromatogr. 2016, 30, 829–836. [Google Scholar] [CrossRef]
- Kabir, A.; Furton, K.G.; Tinari, N.; Grossi, L.; Innosa, D.; Macerola, D.; Tartaglia, A.; Di Donato, V.; D’Ovidio, C.; Locatelli, M. Fabric phase sorptive extraction-high performance liquid chromatography photo diode array detection method for simultaneous monitoring of three inflammatory bowel disease treatment drugs in whole blood, plasma, and urine. J. Chromatogr. B 2018, 1084, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Furton, K.G.; Tartaglia, A.; Sperandio, E.; Ulusoy, H.I.; Kabir, A. An FPSE-HPLC-PDA method for rapid determination of solar UV filters in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1118–1119, 40–50. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; Ulusoy, S.; Sperandio, E.; Piccolantonio, S.; Ulusoy, H.I.; Furton, K.G.; Locatelli, M. FPSE-HPLC-PDA analysis of seven paraben residues in human whole blood, plasma, and urine. J. Chromatogr. B 2019, 1125, 121707. [Google Scholar] [CrossRef] [PubMed]
- Alampanos, V.; Kabir, A.; Furton, K.G.; Samanidou, V. Magnet integrated fabric phase sorptive extraction of selected endocrine disrupting chemicals from human urine followed by high-performance liquid chromatography-photodiode array analysis. J. Chromatogr. B 2021, 1654, e462459. [Google Scholar] [CrossRef]
- Parla, A.; Zormpa, E.; Paloumpis, N.; Kabir, A.; Furton, K.G.; Roje, Z.; Samanidou, V.; Vrcek, I.V.; Panderi, I. Determination of Intact Parabens in the Human Plasma of Cancer and Non-Cancer Patients Using a Validated Fabric Phase Sorptive Extraction Reversed-Phase Liquid Chromatography Method with UV Detection. Molecules 2021, 26, 1526. [Google Scholar] [CrossRef] [PubMed]
- Lioupi, A.; Kabir, A.; Furton, K.G.; Samanidou, V. Fabric phase sorptive extraction for the isolation of five common antidepressants from human urine prior to HPLC-DAD analysis. J. Chromatogr. B 2019, 1118–1119, 171–179. [Google Scholar] [CrossRef]
- Alampanos, V.; Kabir, A.; Furton, K.G.; Samanidou, V.; Papadoyannis, I. Fabric phase sorptive extraction for simultaneous observation of four penicillin antibiotics from human blood serum prior to high performance liquid chromatography and photo-diode array detection. Microchem. J. 2019, 149, e103964. [Google Scholar] [CrossRef]
- Tartaglia, A.; Kabir, A.; D’Ambrosio, F.; Ramundo, P.; Ulusoy, S.; Ulusoy, H.I.; Merone, G.M.; Savini, F.; D’Ovidio, C.; de Grazia, U.; et al. Fast off-line FPSE-HPLC-PDA determination of six NSAIDs in saliva samples. J. Chromatogr. B 2020, 1144, e122082. [Google Scholar] [CrossRef]
- Alampanos, V.; Kabir, A.; Furton, K.G.; Roje, Z.; Vrcek, I.V.; Samanidou, V. Fabric phase sorptive extraction combined with high-performance-liquid chromatography-photodiode array analysis for the determination of seven parabens in human breast tissues: Application to cancerous and non-cancerous samples. J. Chromatogr. A 2020, 1630, e461530. [Google Scholar] [CrossRef] [PubMed]
- Mazaraki, K.; Kabir, A.; Furton, K.G.; Fytianos, K.; Samanidou, V.; Zacharis, C.K. Fast fabric phase sorptive extraction of selected β-blockers from human serum and urine followed by UHPLC-ESI-MS/MS analysis. J. Pharm. Biomed. 2021, 199, e114053. [Google Scholar] [CrossRef] [PubMed]
- Alampanos, V.; Kabir, A.; Furton, K.G.; Samanidou, V. Rapid exposure monitoring of six bisphenols and diethylstilbestrol in human urine using fabric phase sorptive extraction followed by high performance liquid chromatography-photodiode array analysis. J. Chromatogr. B 2021, 1177, e122760. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, A.; Romasco, T.; D’Ovidio, C.; Rosato, E.; Ulusoy, H.I.; Furton, K.G.; Kabir, A.; Locatelli, M. Determination of phenolic compounds in human saliva after oral administration of red wine by high performance liquid chromatography. J. Pharm. Biomed. 2021, 209, e114486. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Tartaglia, A.; D’Ambrosio, F.; Ramundo, P.; Ulusoy, H.I.; Furton, K.G.; Kabir, A. Biofluid sampler: A new gateway for mail-in-analysis of whole blood samples. J. Chromatogr. B 2020, 1143, e122055. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Tartaglia, A.; Ulusoy, H.I.; Ulusoy, S.; Savini, F.; Rossi, S.; Santavenere, F.; Merone, G.M.; Bassotti, E.; D’Ovidio, C.; et al. Fabric-Phase Sorptive Membrane Array as a Noninvasive In Vivo Sampling Device for Human Exposure to Different Compounds. Anal. Chem. 2021, 93, 1957–1961. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Analytes | Matrix | Extraction Technique | Analysis Technique | LOQ | LOD | Ref. |
|---|---|---|---|---|---|---|
| 7-Aminoflunitrazepam | Whole blood | IL-DLLME | LC-ESI-MS/MS | 2 ng/mL | 0.09 ng/mL | [44] |
| Alprazolam | 2 ng/mL | 0.28 ng/mL | ||||
| Bromazepam | 10 ng/mL | 0.67 ng/mL | ||||
| Brotizolam | 2 ng/mL | 1.41 ng/mL | ||||
| Chlordiazepoxide | 50 ng/mL | 4.74 ng/mL | ||||
| Clobazam | 50 ng/mL | 3.49 ng/mL | ||||
| Clonazepam | 2 ng/mL | 0.25 ng/mL | ||||
| Clorazepate | 10 ng/mL | 0.69 ng/mL | ||||
| Clotiazepam | 50 ng/mL | 0.54 ng/mL | ||||
| Cloxazolam | 10 ng/mL | 3.63 ng/mL | ||||
| Diazepam | 50 ng/mL | 2.16 ng/mL | ||||
| Estazolam | 10 ng/mL | 0.40 ng/mL | ||||
| Ethyl loflazepate | 10 ng/mL | 0.94 ng/mL | ||||
| Etizolam | 2 ng/mL | 0.26 ng/mL | ||||
| Flunitrazepam | 2 ng/mL | 0.18 ng/mL | ||||
| Flurazepam | 2 ng/mL | 0.03 ng/mL | ||||
| Loprazolam | 2 ng/mL | 0.003 ng/mL | ||||
| Lorazepam | 2 ng/mL | 0.52 ng/mL | ||||
| Lormetazepam | 2 ng/mL | 0.19 ng/mL | ||||
| Midazolam | 10 ng/mL | 0.50 ng/mL | ||||
| Nitrazepam | 10 ng/mL | 0.61 ng/mL | ||||
| Nordiazepam | 10 ng/mL | 0.69 ng/mL | ||||
| Oxazepam | 50 ng/mL | 1.78 ng/mL | ||||
| Prazepam | 50 ng/mL | 1,83 ng/mL | ||||
| Temazepam | 10 ng/mL | 0.46 ng/mL | ||||
| Triazolam | 2 ng/mL | 0.28 ng/mL | ||||
| Zolpidem | 10 ng/mL | 0.43 ng/mL | ||||
| Zopiclone | 2 ng/mL | 0.33 ng/mL | ||||
| Phenethylamine (2C) | Human plasma | LLE | LC-HRMS | 0.25 ng/mL | 0.1 ng/mL | [45] |
| N-2-methoxybenzyl phenethylamine (NBOMes) | ||||||
| Lysergic acid diethylamide (LSD) | ||||||
| Fentanyl | ||||||
| New Psychoactive Substances (NPS) | Oral fluid | MEPS | UHPLC-MS/MS | 0.015–2.600 ng/mL | 0.005–0.850 ng/mL | [46] |
| 44 opioid-related and analgesic compounds | Postmortem whole blood, urine, liver, brain | SPE | UHPLC-Ion Trap-MS | not reported | 0.1–5 ng/mL | [47] |
| Methadone | Postmortem urine | QuEChERS | GC-MS | 97 ng/mL | 29.1 ng/mL | [10] |
| Amphetamine (AMP) | Human whole blood | UA-DLLME | GC-MS | 40 ng/mL | 10 ng/mL | [48] |
| Methamphetamine (MAMP) | ||||||
| 3,4-Methylenedioxyamphetamine (MDA) | ||||||
| 3,4-Methylenedioxymethamphetamine (MDMA) | ||||||
| Meperidine (MEP) | ||||||
| Methadone (METD) | ||||||
| Ketamine (KET) | ||||||
| Psychoactive Substances (PAS) | Postmortem blood | QuEChERS | UFLC-MS/MS | not reported | 1 ng/mL | [49] |
| (16 antidepressants, 7 antipsychotics and 3 metabolites, norfluoxetine, norsertraline) | ||||||
| 3,4-Methylenedioxymethamphetamine (MDMA) | Postmortem blood | LLME | UFLC-MS/MS | 0.1–1 ng/mL | 0.1–1 ng/mL | [50] |
| 3,4-Methylenedioxyamphetamine (MDA) | ||||||
| 3,4-Methylenedioxyethylamphetamine (MDEA) | ||||||
| N-methoxybenzyl-methoxyphenylethylamine (NBOMes) | ||||||
| Amitriptyline | Postmortem serum, whole blood | QuEChERS | UHPLC-MS/MS | 0.001 µg/mL | 0.0003 µg/mL | [51] |
| Citalopram | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Chlorpromazine | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Clozapine | 0.01 µg/mL | 0.003 µg/mL | ||||
| Fluoxetine | 0.01 µg/mL | 0.003 µg/mL | ||||
| Haloperidol | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Mirtazapine | 0.01 µg/mL | 0.003 µg/mL | ||||
| Nortriptyline | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Olanzapine | 0.05 µg/mL | 0.017 µg/mL | ||||
| Paroxetine | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Quetiapine | 0.01 µg/mL | 0.003 µg/mL | ||||
| Risperidone | 0.005 µg/mL | 0.0017 µg/mL | ||||
| Sertraline | 0.005 µg/mL | 0.0017 µg/mL | ||||
| Venlafaxine | 0.01 µg/mL | 0.003 µg/mL | ||||
| Zolpidem | 0.001 µg/mL | 0.0003 µg/mL | ||||
| Psychoactive drugs and some metabolites | Whole blood | QuEchERS | UPLC-MS/MS | - | - | [52] |
| Barbiturates | Human whole blood | dSPE | GC-MS/MS | 1.1–5.0 ng/mL | 0.36–0.78 ng/mL | [53] |
| Benzodiazepines | 0.21–2.3 ng/mL | |||||
| Tri/tetracyclic antidepressants | 0.32–0.50 ng/mL | |||||
| Drugs of abuse | 1.0–10 ng/mL | 0.20–1.6 ng/mL | ||||
| Phenethylamines | 1.0–5.0 ng/mL | 1.4–4.9 ng/mL | ||||
| Pesticides | 1.0–5.0 ng/mL | 0.09–1.4 ng/mL | ||||
| 10–16 ng/mL | ||||||
| 1.0–5.0 ng/mL | ||||||
| Antidepressants | Postmortem blood | QuEchERS | LC-MS/MS | 10 ng/mL | 10 ng/mL | [54] |
| 2-aminothiazoline-4-carboxylic acid (ATCA) | Synthetic urine | d-μSPE | GC-MS | 10 ng/mL | 5 ng/mL | [55] |
| Bovine blood | 60 ng/mL | 10 ng/mL | ||||
| Amphetamine (AMP) | Oral fluid, urine, serum, sweat, breast milk, hair, and fingernails | SUPRAS | LC-MS/MS | 5–100 ng/mL | not reported | [56] |
| Methamphetamine (MA) | ||||||
| 3,4-Methylenedioxyamphetamine (MDA) | ||||||
| N-ethyl-3,4-methylenedioxyamphetamine (MDEA) | ||||||
| N-methyl-3,4-methylenedioxyamphetamine (MDMA) | ||||||
| Illicit drugs | Urine | DLLME | HPLC-MS | 0.01–0.37 μg/L | 0.006–0.072 μg/L | [57] |
| Synthetic cannabinoids | Oral fluid | DLLME | HPLC-MS/MS | 0.004 ng/mL | 0.002 ng/mL | [58] |
| JWH-200 | 0.013 ng/mL | 0.008 ng/mL | ||||
| AM-694 | 0.009 ng/mL | 0.006 ng/mL | ||||
| JWH-250 | 0.035 ng/mL | 0.021 ng/mL | ||||
| JWH-073 | 0.016 ng/mL | 0.010 ng/mL | ||||
| JWH-018 | 0.030 ng/mL | 0.018 ng/mL | ||||
| JWH-019 | ||||||
| Ketoprofen | Human urine | LLME | HPLC-UV | not reported | 15 μg/L | [59] |
| Diclofenac | 44 μg/L | |||||
| Nortriptyline | Human breast milk, urine | DLPME | IMS | 5.0 μg/L | 1.9 μg/L | [60] |
| Sertraline (SER) | Human plasma | HS-SPME | IMS | 0.26 µg/mL | 0.08 µg/mL | [61] |
| Imipramine (IMI) | 0.12 µg/mL | |||||
| Alprazolam (ALP) | 0.39 µg/mL | 0.27 µg/mL | ||||
| 0.89 µg/mL | ||||||
| Chlorpromazine | Human plasma | SPE | LC-MS/MS | 13.17 ng/mL | 3.95 ng/mL | [62] |
| Haloperidol | 1.19 ng/mL | 0.36 ng/mL | ||||
| Levomepromazine | 4.99 ng/mL | 1.50 ng/mL | ||||
| Olanzapine | 2.89 ng/mL | 0.87 ng/mL | ||||
| Risperidone | 4.59 ng/mL | 1.378 ng/mL | ||||
| Sulpiride | 7.04 ng/mL | 2.11 ng/mL | ||||
| Antipsychotic | Plasma | LLE | LC-MS/MS | not reported | not reported | [63] |
| Anastrozole | Human whole blood, plasma, urine | FPSE | HPLC-PDA | 0.05 μg/mL | 0.02 μg/mL | [64] |
| Letrozole | ||||||
| Exemestane | ||||||
| Ketamine (KET) | Human plasma | PPE | HPLC-MS/MS | 20 ng/mL | 10 ng/mL | [65] |
| Midazolam (MDZ) | 10 ng/mL | 0.5 ng/mL | ||||
| Dehydro-norketamine (DHNK) | 320 ng/mL | 300 ng/mL | ||||
| Nor-ketamine (NK) | 470 ng/mL | 410 ng/mL | ||||
| Midazolam 1-hydroxy midazolam (1HMDZ) | 150 ng/mL | 100 ng/mL | ||||
| Atenolol | Human urine samples | tCap-μEx | CE | 3.27 ng/mL | 1.13 ng/mL | [66] |
| Metoprolol | 2.10 ng/mL | 0.75 ng/mL | ||||
| Tacrolimus | Human whole blood | SPME | MOI-MS/MS | 0.8 ng/mL | 0.3 ng/mL | [67] |
| Sirolimus | 0.7 ng/mL | 0.2 ng/mL | ||||
| Everolimus | 1 ng/mL | 0.3 ng/mL | ||||
| Cyclosporine A | 0.8 ng/mL | 0.3 ng/mL | ||||
| Ketoconazole | Human plasma, Urine | FPSE | HPLC-PDA | 0.1µg/mL | 0.03 µg/mL | [68] |
| Terconazole | ||||||
| Voriconazole | ||||||
| Bifonazole | ||||||
| Clotrimazole | ||||||
| Tioconazole | ||||||
| Econazole | ||||||
| Butoconazole | ||||||
| Miconazole | ||||||
| Posaconazole | ||||||
| Ravuconazol | ||||||
| Itraconazole | ||||||
| Ketamine (KET) | Human serum, urine, feces | LLE | LC-MS/MS | 0.1 ng/mL | not reported | [69] |
| Nor ketamine (n-KET) | ||||||
| Dehydronorketamine (DHNK) | ||||||
| Hydroxynorketamine (HNK) | ||||||
| Hydroxyketamine (HK) | ||||||
| Atenolol | Plasma | LPME-DESs | GC-MS | 0.645 ng/mL | 0.195 ng/mL | [70] |
| Propranolol | 0.435 ng/mL | 0.130 ng/mL | ||||
| Metoprolol | 0.692 ng/mL; | 0.205 ng/mL | ||||
| Nortriptyline | Human urine, dried blood spot | μ-EME | CE-UV | not reported | 5–28 ng/mL | [71] |
| Papaverine | ||||||
| Loperamide | ||||||
| Haloperidol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ovidio, C.; Bonelli, M.; Rosato, E.; Tartaglia, A.; Ulusoy, H.İ.; Samanidou, V.; Furton, K.G.; Kabir, A.; Ali, I.; Savini, F.; et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations 2022, 9, 18. https://doi.org/10.3390/separations9010018
D’Ovidio C, Bonelli M, Rosato E, Tartaglia A, Ulusoy Hİ, Samanidou V, Furton KG, Kabir A, Ali I, Savini F, et al. Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses. Separations. 2022; 9(1):18. https://doi.org/10.3390/separations9010018
Chicago/Turabian StyleD’Ovidio, Cristian, Martina Bonelli, Enrica Rosato, Angela Tartaglia, Halil İbrahim Ulusoy, Victoria Samanidou, Kenneth G. Furton, Abuzar Kabir, Imran Ali, Fabio Savini, and et al. 2022. "Novel Applications of Microextraction Techniques Focused on Biological and Forensic Analyses" Separations 9, no. 1: 18. https://doi.org/10.3390/separations9010018