1. Introduction
The availability of high-quality genetic material is necessary in paraclinical settings for reliable diagnosis, prognostics, and treatment and is especially relevant in the era of precision medicine. Validated, reliable purification protocols ensuring high RNA and/or DNA amounts for molecular testing are important, especially in clinical contexts where the patient material is often both scarce and unique and where the assay result impacts patient diagnosis and treatment.
An RNA/DNA purification protocol consists of four steps: effective tissue disruption, denaturation of nucleoprotein complexes, inactivation of nucleases, and removal of contaminants. Methods allowing nucleic acid extraction can be divided into solution-based, which is currently rarely used, column-based or involving magnetic separation [
1,
2]. Current trends in molecular biology include increasing automation of the extraction process and options such as all-in-one commercial kits, allowing simultaneous extraction of RNA and DNA often along with protein [
1].
Numerous high-quality DNA and RNA purification kits manufactured by different providers and suitable for various types of starting material are currently available on the market. Studies comparing their performance have previously been attempted, and they rarely demonstrate that one product clearly outperforms others in the panel [
3,
4,
5,
6]. However, only a few providers offer the option of simultaneous extraction of DNA and RNA from the same fraction of starting material. This solution may be advantageous when working with small amounts of starting material or to account for possible sample heterogeneity and ensure that both DNA and RNA come from an area with similar cell composition (as underlined by Pena-Llopis and Brugarolas, [
7]).
It should also be stressed that working with clinical specimens presents additional challenges in terms of standardization, as compared to, for instance, purification of DNA/RNA from pure cell lines. In the latter case, it is theoretically possible to relate the actual nucleic acid yield to the predicted content. In the case of tissue samples, DNA/RNA extraction efficiency often varies substantially, which may result from the properties of tissue (fibrous, with low cellularity or high fat content) but also from the clinical state (ischemia, apoptosis, necrosis) [
8,
9,
10]. We have also observed this in our experimental and clinical routine setting and have found that especially breast and testis tissue samples may be challenging in regard to nucleic acid extraction. Thus, our objective was to improve the nucleic acid extraction and ensure sufficient amounts of high-quality RNA and DNA, irrespective of the tissue of origin and preferably using the same fraction of starting material for simultaneous extraction of both molecules. The procedure should be easy to perform as a standard laboratory procedure and does not present a health hazard. We achieved this by identifying an extraction kit that would perform well in challenging samples.
3. Discussion
The availability of a good quality biological material is a prerequisite of obtaining credible results in molecular diagnostics. To ensure reliable data, both RNA and DNA should be intact (undegraded), clean (i.e., devoid of potential inhibitors of downstream processes) [
12], and present in sufficient amounts.
Choice of tissue disruption method and the appropriate lysis buffer is crucial for the first steps of nucleic acid purification. Tissue homogenization must be thorough to ensure high yield whilst not damaging the material. Nouvel et al. compared various tissue disruption techniques and found that they may affect not only RNA yield but also some of the RNA integrity markers, with 28S rRNA being especially sensitive to mechanical shearing. We chose a relatively gentle homogenization method, which was sufficient for the biospecimens included in our study. However, since skin is reportedly difficult to disrupt, more harsh methods (such as bead-beating) may be necessary for this sample type [
10,
13].
Tissue disruption should also be fast to prevent enzymatic degradation of nucleic acids by nucleases. RNA digestion by RNases is an especially common pitfall. Prolonged time until freezing or until placing in a solution containing RNase inhibitors, designed either to preserve (RNAlater) or to lyse the sample (RLT, QIAzol), may significantly affect RNA integrity. Moreover, if the perfusion of the preserving/lysing solution is not fast enough (like in dense samples rich in connective tissue), RNA degradation can be more pronounced [
10].
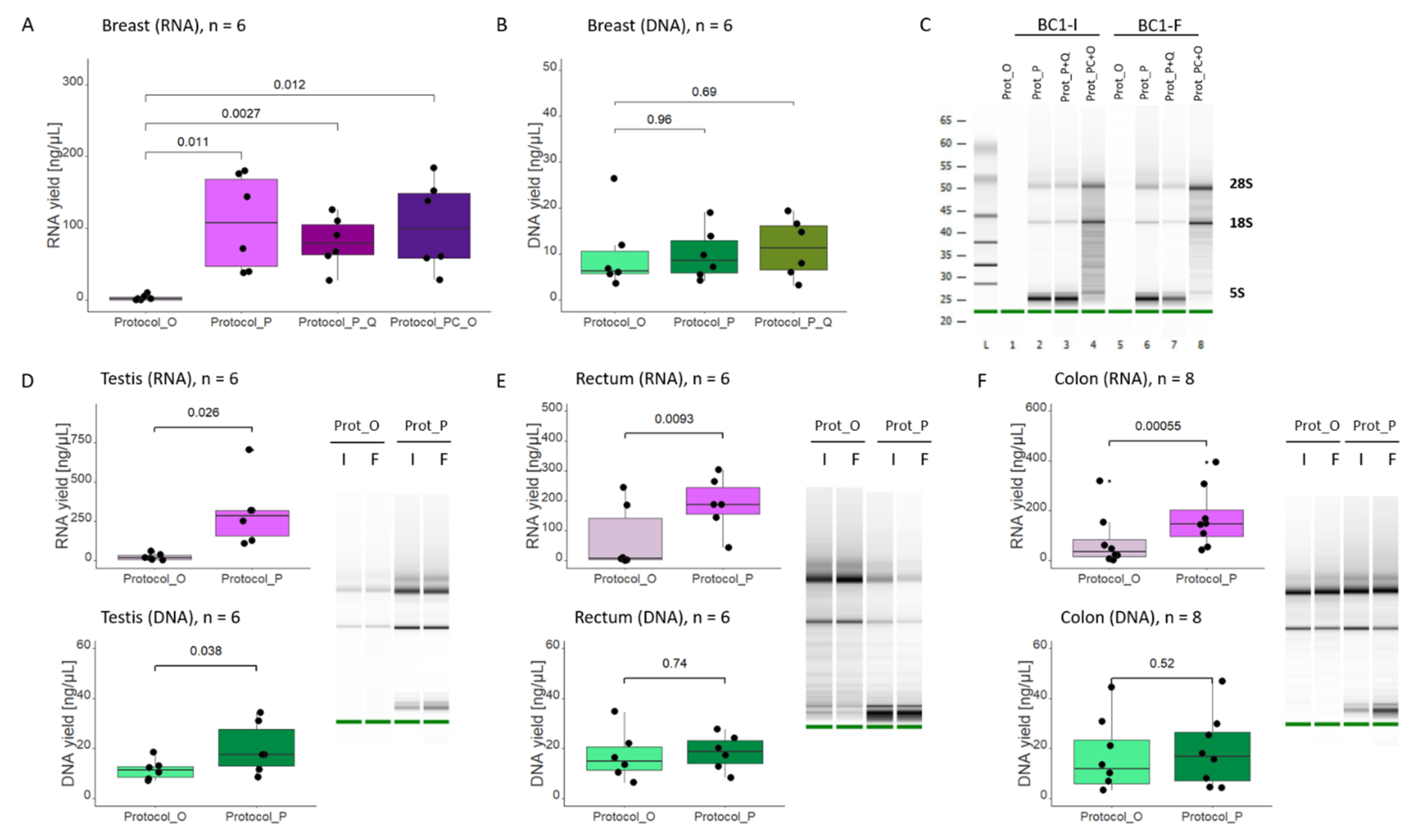
One of the strategies for successful RNA isolation, especially from RNase-rich tissues, is the use of proteinase K. Proteinase K may be added either at the initial steps, directly to the cell/tissue homogenate, where its role is to facilitate sample digestion (for example, when working with highly fibrous tissue) or, as in our case (
Protocol_P, #80224), to the lysate devoid of tissue debris. In such a scenario, the role of proteinase K is to inactivate nucleases that could degrade RNA (or DNA) during purification and to digest proteins, thus freeing RNA from RNA-protein complexes and decreasing sample contamination [
14]. In our case, using an extraction kit containing proteinase K ensured high RNA yield, even from samples regarded as “difficult”. Another approach that, in our experimental setting, ensured high RNA yield, was tissue disruption in QIAzol reagent, followed by phenol-chloroform extraction and subsequent processing of the aqueous phase with the #80204 kit (
Protocol_PC+O). QIAzol ensures efficient tissue lysis, though the method has two major drawbacks. First, phenol-based reagents (such as TRIzol or QIAzol) are toxic, and working with them presents a certain health hazard. Then, unlike the AllPrep protocol, phenol-chloroform extraction only supports RNA isolation. This would mean the necessity of using two different methods, depending on the desired outcome. Therefore, instead of resorting to phenol-chloroform extraction, we settled on using
Protocol_P (#80224), which also resulted in high yield of RNA and moreover allowed simultaneous extraction of both RNA and DNA.
At the same time, we observed high amounts of low molecular weight rRNA being co-purified when using Protocol_P (#80224). The extraction is facilitated by ethanol concentration and buffer composition (FRN instead of RW1), not using proteinase K. Therefore, the desired outcome would be to optimize the Protocol_O (#80204) so that it includes a proteinase K digestion step, without co-extracting small RNA. To our knowledge, so far, the chemistry of the RLT Plus buffer does not allow this (QIAGEN, personal communication).
The suitability of extracted RNA and DNA for downstream applications is regarded in terms of integrity and purity. Assessment of RNA integrity is commonly made using the RIN parameter, designed for unambiguous and user-independent interpretation of RNA quality. It considers the whole electropherogram, rather than 18S and 28S rRNA peaks alone, and is more robust to sample dilution and inter-instrument variability than the 28S/18S ribosomal ratio [
15]. However, presence of unexpected peaks (such as a very high peak is 5S region, as we observed) may impede RIN calculation. We discussed this finding with both QIAGEN and Agilent customer service. They supported our conclusions and agreed that the flat baseline between 5S and ribosomal peaks suggests that the observed peak is due to the presence of small RNA, rather than RNA degradation (QIAGEN and Agilent customer service, personal communication). In such a case, the 28S/18S ribosomal ratio is the parameter of choice, and a significant positive correlation between ribosomal ratio and RIN values has been shown previously [
10].
It must be stressed that the Bioanalyzer images, unless using specifically mRNA-enriched or ribo-depleted samples, only give information about the integrity of ribosomal RNA. mRNA constitutes around 4% of total RNA [
16] and is not visible on Bioanalyzer electropherograms. Therefore, Bioanalyzer data only allow to infer about mRNA integrity based on the rRNA quality, and RINs serve as a surrogate parameter. Of note, only samples with a RNA concentration exceeding 25 ng/µL can be accurately measured using the Bioanalyzer [
15], though samples with lower RNA content can potentially still be used in downstream applications. In our study, we successfully performed RT-qPCR on breast and testis samples with concentrations below 10 ng/µL (as low as 1.3 ng/µL or 8 ng RNA per cDNA reaction), for which Bioanalyzer electropherograms were blank.
Analysis of Bioanalyzer images provided information both about RNA integrity and RNA size distribution. The peak/band of small RNA in the 5S region of the Bioanalyzer electrogram constituted from 20 to even 80% of the total RNA profile. We were primarily interested in gene expression assays and therefore in obtaining good quality mRNA defined as fragments composing several hundred nucleotides. Therefore, our concern was that the high share of small RNA would interfere with the assay and, at best, only make our concentration measurement (by Qubit or NanoDrop) imprecise. When we compared the Ct values for samples isolated either with Protocol_O or Protocol_P, we observed that Protocol_P samples tend to have higher Ct values, which would confirm our hypothesis that part of the Qubit/NanoDrop measurement comes from small RNA. However, the differences were typically around 1 Ct, which means that the Qubit/NanoDrop measurement still gives a relatively good estimate of the amount of mRNA starting material.
Nucleic acid purity is another factor that may influence their performance in downstream experiments. Several classes of substances may potentially act as PCR inhibitors (reviewed by Shrader et al.; [
12]). Contaminants may originate from the specimen itself (polysaccharides, bile salts, various proteins) or be introduced during purification (phenols). Two parameters (purity ratios) are commonly used to estimate the presence of contaminants: relative absorbance at 280 nm (260/280 ratio) and at 230 nm (260/230 ratio). The first parameter informs about protein contamination (optimal values are around 1.8 for DNA and 2 for RNA). The second parameter (optimal values are in the range of 1.8–2.2) may be affected by the presence of various organic compounds, such as peptides, carbohydrates, or phenols [
17]. An example of a compound absorbing very strongly at 230 nm is guanidine thiocyanate, and it is present in high amounts in lysis buffers (including RLT) or extraction reagents such as TRIzol or QIAzol. Guanidine thiocyanate is therefore regarded as the main culprit responsible for low 260/230 ratios; several studies report that even very low 260/230 ratios do not affect performance of downstream applications, including RT-PCR, multiplex PCR, NGS, and microarray [
18,
19,
20].
The AllPrep system launched by QIAGEN allows simultaneous extraction of DNA and RNA from the entire sample. Another commonly used option is to divide the sample or already purified nucleic acids into two and process them separately (for example, by adding RNase to one and DNase to the other part). However, half of the material is lost. The protocol implemented by QIAGEN allows separation into DNA and RNA fractions, based on selective properties of AllPrep DNA Mini Spin Columns. The separation is not perfect, and the genomic DNA, albeit in small amounts (Ct values > 34), can still be detected in the RNA fraction (see
Figure 3D).
Furthermore, we demonstrated that the AllPrep DNA/RNA/miRNA Universal kit has a clear advantage over the AllPrep DNA/RNA Mini kit in terms of RNA yield, which most likely can be ascribed to the proteinase K digestion step.
In summary, we successfully improved our extraction procedure, demonstrating that good quality RNA and DNA may be purified simultaneously, even from samples previously regarded as problematic, and avoiding the health hazard posed by using phenol-chloroform extraction. Our new protocol of choice, involving the use of the AllPrep DNA/RNA/miRNA Universal kit #80224, ensures high amounts of broad-range molecular weight RNA, and though 5S RNA peak obscures Bioanalyzer analysis, Qubit/NanoDrop measurements still provide a good estimate of the amount of starting material for downstream analyses such as RT-qPCR.
4. Materials and Methods
4.1. Human Tissue Samples
The material used in this study consisted of fresh-frozen tissue cancer specimens, obtained for diagnostic purposes by the Department of Pathology, Herlev and Gentofte Hospital, Denmark, and stored at the Danish CancerBiobank (part of the Bio- and Genome Bank Denmark; RBGB). All specimens were collected and handled according to strict standard operating procedures. Tissue samples from the following organs were included: breast, colon, testis, rectum, uterus, adrenal gland, and kidney. Samples were snap-frozen upon collection either in dry-ice cooled isopentane (VWR Chemicals, Solon, OH, USA) or using a FlashFREEZE benchtop unit (Milestone Medical, Sorisole, Italy); two specimens, frozen according to two different methods, were available from each subject.
When comparing extraction protocols, each specimen was divided into smaller samples, which were then processed in parallel.
The overview of all samples, including the tissue of origin and the analysis employed, is gathered in
Table 5.
4.2. Nucleic Acid Extraction
Altogether, for comparative purposes, we implemented four purification protocols, referred to as Protocol_O, Protocol_P, Protocol_P+Q, and Protocol_PC+O, as described below.
In all protocols, apart from the guanidine thiocyanate-phenol-chloroform (PC+O) method, tissue was disrupted in RLT Plus buffer (QIAGEN, Hilden, DE, USA). Briefly, fragments of fresh-frozen specimens (not exceeding 3 mm3 or 20 mg) were cut into smaller pieces with a scalpel, immediately immersed in 200 µL RLT Plus buffer containing 0.1% v/v β-mercaptoethanol, and disrupted using a pestle (Thermo Fisher Scientific, Waltham, MA, USA). When processed, tissue specimens were kept on dry ice util lysis. Visually, all samples had similar size.
After initial disruption, 400 µL RLT Plus Buffer was added, and samples were lysed for an additional 5 min before continuing with one of the two commercial kits. The different protocols employed in our settings are described below:
Protocol_O—(originally implemented protocol). After tissue disruption in RTL Plus Buffer, samples were centrifugated at maximum speed for 3 min, and the supernatant was further processed as instructed by the manufacturer using the AllPrep DNA/RNA Mini Kit, #80204 kit (QIAGEN).
Protocol_P (proteinase K)—Analogous to Protocol_O, except for implementing a different kit, namely AllPrep DNA/RNA/miRNA Universal kit #80224 (QIAGEN) containing proteinase K and an on-column DNAseI digestion step, as instructed by the manufacturer.
Protocol_P+Q (proteinase K + QIAshredder)—Analogous to Protocol_P, with the only difference being the use of QIAshredder columns (QIAGEN, cat. No. 79656). Immediately after lysis in RLT Plus Buffer, samples were centrifugated in QIAshredder columns instead of 1.5 mL Eppendorf tubes.
Protocol_PC+O—tissue specimens were disrupted in 200 µL QIAzol reagent (QIAGEN) instead of RLT Plus Buffer. After tissue disruption, 800 µL of QIAzol was added to the sample (up to 1 mL) and mixed with 200 µL chloroform. The tube was shaken vigorously for 15 s and incubated at room temperature for 2–3 min, followed by centrifugation of the lysate at 12,000 g for 15 min. Then, the upper, aqueous phase was transferred into a fresh tube, and the isolation continued using AllPrep DNA/RNA Mini Kit, #80204 kit (QIAGEN; Protocol_O), as instructed by the manufacturer. Elution in all protocols was done with 24 µL water for RNA extraction and 100 µL EB buffer for DNA extraction.
The protocol names we use throughout the text are for readers’ convenience, to avoid repeating product names or using unintuitive product catalogue numbers. However, it should be noted that Protocol_O and Protocol_P, apart from the method suggested for tissue disruption herein (pestle), refer to original QIAGEN procedures.
4.3. Evaluation of DNA/RNA Quantity and Purity
Nucleic acid concentration was quantified by a Qubit® 2.0 Fluorometer with Qubit™ dsDNA HS Assay Kit for DNA and Qubit™ RNA HS Assay Kit for RNA and with NanoDrop™ One. Sample purity (260 nm/280 nm and 260 nm/230 nm absorbance ratios) was evaluated by NanoDrop™ One (all from Thermo Fisher Scientific). All samples were measured in a 1:200 dilution according to the manufacturer’s procedures, leading to a limit of quantification of 4 ng/µL for RNA and 0.5 ng/µL for DNA. In a few samples, a lower dilution was used in order to detect lower levels.
4.4. Analysis of RNA Integrity
RNA integrity was evaluated by microcapillary electrophoresis using an Agilent 2100 Bioanalyzer with an Agilent RNA 6000 Nano Kit (Agilent technologies, Santa Clara, CA, USA). One microliter of undiluted sample was subjected to the analysis according to manufacturer’s instructions. RNA integrity was expressed as 28S/18S ribosomal peak ratio. The 28S/18S peak ratio and the percentage share of 5S peak were calculated using Bioanalyzer 2100 Expert software (Agilent).
4.5. Reverse Transcription—Quantitative Polymerase Chain Reaction (RT-qPCR) on RNA
RNA was transcribed into cDNA using the AffinityScript QPCR cDNA Synthesis Kit and oligo(dT) primers according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA, USA). For each sample pair (purified with different protocols), the amount of total RNA used as input for cDNA synthesis was identical. Real-time measurement of mRNA levels was performed with the Stratagene 3005P qPCR System (Agilent Technologies) using TaqMan® Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) specific for GAPDH (assay ID: Hs02758991_g1) and ACTB (assay ID: Hs99999903_m1). Threshold cycle (Ct) values for each sample were collected and compared between protocols.
4.6. Quantitative Polymerase Chain Reaction (qPCR) on DNA
DNA performance in qPCR was estimated using BRAF mutation detection kits (EntroGen, Woodland Hills, CA, USA) and analyzed on the ABI 7500 Fast Real Time PCR system.
Briefly, four 10-fold DNA serial dilutions were prepared (40, 4, 0.4, and 0.04 ng DNA per sample), and the qPCR reaction was carried out according to manufacturer’s protocol (EntroGen). Linearity and efficiency of the PCR amplification reactions were evaluated as follows. The lm (linear model) function (package stats, R version 3.6.1) was used to find the linear relation between the DNA input (log10 of DNA concentration) and Ct values. Since no mutations were detected, we collected the signal for internal control (housekeeping gene). Two parameters: coefficient of determination (R2), measuring the goodness-of-fit in a linear regression, and the slope were extracted. The slope was subsequently used to calculate the PCR reaction efficiency (E), using the formula: E = −1 + 10(−1/slope).
4.7. Genomic DNA Contamination Analysis
The BRAF mutation detection kit (EntroGen) was used for analysis of the presence of contaminating DNA in RNA samples. The input material consisted of purified RNA samples and amounts of total RNA corresponding to amounts used for cDNA synthesis (see RT-qPCR).
4.8. Statistical Analysis
Samples extracted with Protocol_O were compared with corresponding samples extracted with Protocol_P or with one of the protocols used in the first part of the project (protocols P+Q and PC+O) using the two-sided t-test or Wilcoxon signed rank (for non-Gaussian distributions) with a significance level alpha = 0.05. Data distributions were tested for normality using a quantile–quantile plot. Correlation between two variables was estimated by Pearson correlation, and two parameters, correlation coefficient and p-value, were collected. Statistical analysis and graph preparation were performed in R programming language (version 3.6.1).









{kind=link}
{kind=link}
{kind=link}
{kind=link}