1. Introduction
Since the end of 90s, scientists have warned of the danger that sewage containing antineoplastic compounds can pose adverse effect on human health and well-being and demanded regular monitoring of these chemical entities [
1]. These pharmaceutical compounds enter into the sewage mainly after patient’s treatment [
2,
3,
4], and they are known to be carcinogenic, mutagenic, teratogenic and/or fetotoxic [
2,
5,
6]. In addition, many authors have demonstrated that several antineoplastic compounds do not degrade in classical wastewater treatment plants (WWTPs) [
7,
8,
9], not even after passing through the post-treatment regimens [
10] and may generate toxic degradation products [
1] that finally end up in rivers or seas. Their long-term effects on non-target organisms and their environmental significance are not clear [
11], although some researchers have published lowest observed effect concentration in eco-toxicological tests close to the concentrations detected in effluent from WWTPs [
12]. For all of the above, they are considered one of the most toxic pharmaceutical compounds used nowadays [
13], and must be monitored to ensure a safe ecosystem.
Anthracyclines are an important group of antineoplastic compounds that act by preventing cell division or disrupting DNA. This group of antineoplastic compounds is mainly used in the therapeutic fight against acute leukaemia, also as a combination therapy for lymphoma and for solid tumours [
14].
Different authors have optimised methodologies to determine them at trace levels in sewage by applying solid phase extraction (SPE) procedures that usually requires large sample volumes and long overall sample preparation times. The anthracyclines commonly determined include doxorubicin (DOX), daunorubicin (DAU) and epirubicin (EPI), but other anthracyclines like doxorubicinol (DOXOL) have been also studied.
Recoveries achieved for these compounds using SPE is variable, despite the fact that almost all the authors used the same cartridge (Oasis HLB). Gomez- Canela et al. [
15] achieved good relative recoveries: 100% for DOX, 73% for DAU and 104% for EPI. However, most of them, the recovery was ranging from 57–75% [
16,
17,
18,
19]. Adapting a previous methodology [
15], Franquet-Griell et al. [
20] obtained a recovery ranging from 48–75%, but using in river water instead of effluents of WWTPs. In these works, a LC-MS/MS system was used for the determination of the analytes. In another work, Gomez-Canela et al. [
21] obtained a recovery of 37% for EPI using SPE before LC–Orbitrap–MS, attributing the low recovery to the poor extraction performance of the Oasis HLB extraction sorbent. All methodologies, except Rabii et al. [
19], used the same cartridge and obtained a range of relative recovery values for these compounds between 37% to 104%.
Furthermore, SPE is not a very selective technique, and while it preconcentrates the analytes of interest, it also simultaneously preconcentrates the matrix interferents. Microextraction techniques can overcome the drawbacks of SPE extraction. These processes have the advantage of reducing organic solvent usage, lowering sample volume and extraction time, which are beneficial to both, the environment and the researchers. Selective extraction and preconcentration of highly polar analytes like anthracyclines from an overly complex sample matrix such as sewage water pose a great analytical challenge. Among the available extraction and microextraction techniques, fabric phase sorptive extraction (FPSE) is the only sample preparation technique that, by design, combines the exhaustive extraction principle (primary extraction mechanism in SPE) and the equilibrium extraction principle (primary extraction mechanism in SPME). Due to the simultaneous exploitation of two major extraction principles, high primary contact surface area of FPSE membrane and sponge like porous architecture of sol-gel sorbents, FPSE provides high absolute analyte recovery of polar, medium polar and nonpolar analytes with a very fast extraction kinetics. Unlike other sample preparation techniques, the sorbents in FPSE are chemically bonded to the fabric substrate that allows the exposure of the FPSE membranes to any organic solvent to maximize the analyte recovery after the extraction process. FPSE has also eliminated sample pretreatment processes to remove matrix interferents such as filtration and post treatment processes such as solvent evaporation and sample reconstitution from the sample preparation workflow. In order to successfully carry out the extraction and monitoring of antineoplastic compounds, it is important to develop rapid, cheap and sensitive methods that allow their appropriate determination [
22].
Most of the authors have used liquid chromatography tandem mass spectrometry (LC-MS/MS) as the separation and determination technique, which allows to measure very low concentrations, but it is expensive and can be strongly influenced by the matrix effect that can produce, in most cases, a significant suppression of the analyte signal. For this reason, it is a parameter that must be evaluated; however, few authors have done so. In this sense, and regarding to anthracyclines, Yin et al. [
16] obtained a range of signal suppression in sewage from 6–78%. On the other hand, Martín et al. [
17] had reported practically no matrix effect, ranging from −5 to 20%. Franquet-Griell et al. [
20] obtained also a low matrix effect (between −28 and 6%) but, in this case, it may be attributed to the studied matrix (river water). Rabii et al. [
19] suffered also a significant suppression of the signal of 78% for EPI using on-line SPE and Gomez-Canela et al. [
21] obtained a recovery of 44% when EPI was injected directly into the LC–Orbitrap–MS equipment. They explained the low recovery due to ion suppression or poor water stability, that is, matrix effect.
To the best of our knowledge, only two works have used other extraction techniques different from SPE for anthracyclines determination. Gouveia et al. [
23] developed a methodology based on liquid-liquid extraction (LLE) followed by LC-MS for the extraction of 14 antineoplastic compounds including DOX. They obtained a recovery ranging from 105–132% for DOX in sewage and an instrumental detection limit (IDL) of 6.47 ng·L
−1. Souza et al. [
24] have developed a microextraction methodology for this family of compounds, based on dispersive liquid-liquid microextraction (DLLME) and HPLC with fluorescence detection. The compounds analysed were Irinotecan, DOX, DAU and EPI. They obtained a recovery range from 74.28–90.01% in hospital effluents without almost any matrix effect, obtaining a limit of quantification (LOQ) of 1 µg·L
−1.
In this work, we have optimised and applied FPSE technique [
25] to the extraction of three commonly used anthracyclines (DOX, DAU and EPI) prior to their determination by ultra-high performance liquid chromatography with fluorescence detection. A systematic optimisation of the most suitable fabric phase sorptive extraction sorbent chemistry and extraction conditions through a statistical design of experiment (DoE) was carried out.
2. Materials and Methods
2.1. Materials and Reagents
Sol-gel sorbent-coated FPSE membranes used in the current study were prepared in Florida International University, Miami, Florida, USA. All chemicals, reagents, solvents, organic polymers and sol-gel precursors used in the project were of the highest quality that can be acquired commercially. Methyltrimethoxysilane (MTMOS), trifluoroacetic acid (TFA), acetone, polytetrahydrofuran (PTHF), poly (ethylene glycol) 300, UCON HTF14 and dichloromethane were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cyanopropyl trimethoxysilane, poly(caprolactone-dimethylsiloxane-caprolactone) block polymer and tetramethyl orthosilicate were purchased from Gelest Inc. (Morrisville, PA, USA). Sodium hydroxide, hydrochloric acid and CW20M were purchased from Fisher Scientific (Pittsburg, PA, USA). Deionized water and acetonitrile were obtained from Fisher Scientific (Pittsburg, PA, USA). Muslin cotton fabric (100% cellulose) was purchased from Jo-Ann Fabric (Miami, FL, USA). An Eppendorf Centrifuge 5415 R (Eppendorf North America Inc., Hauppauge, NY, USA) was used to remove unwanted and interfering micro particles from the sol solutions prior to sol-gel coating on the fabric substrate used to create fabric phase sorptive extraction membrane. A Philips XL30 Scanning Electron Microscope equipped with an EDAX detector was used to obtain SEM images. A Barnstead NANOPure Diamond (Model D11911) deionized water system (Barnstead Inc., Dubuque, IA, USA) was used to obtain deionized water (18.0 MΩ).
HPLC-grade methanol (MeOH), MS- grade acetonitrile (ACN), formic acid (F.A.) and ammonia were all obtained from Panreac Quimica (Barcelona, Spain). Ultrapure water used was provided by a Milli-Q system (Milli-pore, Bedford, MA, USA). Cytostatics compounds (DOX, DAU and EPI) were purchased from Cymit-Química (Barcelona, Spain). Stock solutions containing 500 mg·L
−1 (DAU and EPI) and 1000 mg·L
−1 (DOX) were prepared by dissolving the compound in MeOH. The solutions were stored in glass-stoppered bottles at −20 °C in the dark. Working standard solutions were prepared daily. The studied compounds and their properties are shown in
Table 1.
Seven different fabric phase sorptive extraction membranes coated with different sorbents were used in this study: sol-gel CW20M, sol-gel PEG300, sol-gel UCON, sol-gel PTHF250, sol-gel Silica, sol-gel CN-CW20M and sol-gel PCAP-PDMS-PCAP. The main features of these sorbents are presented in
Table 2 and a photograph of the appearance of the fabrics used is shown in the
Figure 1.
2.2. Creation of Sol-Gel Sorbent Coated Fabric Phase Sorptive Extraction Membrane
Hydrophilic 100% cellulose Muslin cotton fabric was used as the substrate for sol-gel sorbent coating. Commercial cotton cellulose fabric contains different surface finishing chemicals as well as other additives to improve the overall appearance of the fabric. However, these chemicals conceal a significant portion of surface hydroxyl functional group and need to be removed to maximize the surface hydroxyl group prior to the sol-gel sorbent coating. The detailed surface treatment process of cellulose fabric is presented elsewhere [
26,
27].
As the target analytes are highly polar (
Table 1), seven different sol-gel sorbents possessing high to moderate polarity were synthesized and evaluated. The sol-gel sorbents include sol-gel CW20M, sol-gel PEG300, sol-gel UCON, sol-gel PTHF250, sol-gel Silica, sol-gel CN-CW20M and sol-gel PCAP-PDMS-PCAP. The formulations of sol-gel CW20M, sol-gel PEG300, sol-gel PTHF250 and sol-gel PCAP-PDMS-PCAP sorbents are described elsewhere [
28,
29]. Sol-gel Silica sorbent coating was prepared using tetramethyl orthosilicate as the only sol-gel precursor/polymer. Sol-gel UCON and sol-gel CN-CW20M sorbent coatings were prepared using methyl trimethoxysilane and 3-cyanopropyl trimethoxysilane, respectively. The major building blocks of the sol-gel sorbents are presented in
Table 2. The detail procedure of sol solution preparation and subsequent coating and post-treatment procedures are presented elsewhere [
29] Briefly, the sol solution was prepared by the sequential addition of 5 g organic/inorganic polymer, 10 mL acetone: methylene chloride (1:1
v/
v), 5.0 mL methyltrimethoxysilane and 2.0 mL trifluoroacetic acid (containing 5%
v/
v water). The sol solution was vortexed vigorously after adding each of the ingredients to ensure that the resulting solution becomes homogeneous and particle free. The sol solution was then subjected to sonication to remove any trapped air bubbles. Finally, the sol solution was transferred into a 30 mL amber reaction vessel and a 10 cm × 5 cm piece of clean and treated cotton fabric was gently immersed into the sol solution. The sol solution was allowed to crate the sol-gel sorbent coating on the fabric substrate for 4 h at room temperature.
At the end of the sol-gel sorbent coating process, the sorbent coated fabric was removed from the reaction vessel and was stored in a desiccator overnight. Subsequently, the sol-gel sorbent coated fabric was rinsed with acetone: methylene chloride (1:1 v/v) under sonication for 30 min. The sol-gel sorbent coated membrane was then air dried for 1 h and was cut into 1 cm × 1 cm pieces. The FPSE membranes were then stored in an air-tight container until their application in sample preparation.
2.3. Instrumentation and Chromatographic Conditions
Used instrument include an ultra-high performance liquid chromatography system equipped with an ACQUITY Quaternary Solvent Manager (QSM), an autosampler, a column manager and a fluorescence detector (UHPLC-FD) from Waters (Barcelona, Spain). The excitation and emission wavelengths selected were 470 and 550 nm for all compounds.
The analytical column was a 50 mm × 2.1 mm, Phenomenex Luna Omega column with a particle size of 1.7 μm (Phenomenex, Madrid, Spain) operating at room temperature. The sample volume injected was 10 µL, and the analyte separation was carried out using water (A) and ACN (B) both with 0.1% (v/v) of formic acid at a flow rate of 0.4 mL∙min−1 in gradient mode. The gradient started at a 70:30 (v/v) mixture of A: B and remained in this composition for 0.3 min. Then, it was changed to 15:85 (v/v) in 0.7 min and remained in this composition from 1 to 1.4 min. After that, the percentage of A was decreased again to 0:100 (v/v) in 0.1 min and remained in this composition for 0.5 min. Mobile phase A was increased from 2.0 min to 3. 5 min to achieve the initial composition 70:30 (v/v) A: B and remained in this composition for 1.5 min to equilibrate the pressure, achieving the chromatographic separation in 5 min.
2.4. Fabric Phase Sorptive Extraction Procedure
Before analysis, FPSE membranes were preconditioned in 2 mL of a mixture MeOH/ACN (50:50, v/v) for 5 min and then in 2 mL of Milli-Q water for another 5 min. After that, the FPSE membrane was placed in contact with the 20 mL of the sample (adjusted at pH = 3) for 15 min and stirred at 1000 rpm. After extraction, analytes were eluted with 1 mL of MeOH/ACN (50:50, v/v) + 10% F.A. for 4 min. The elution process was repeated and then the solvent was collected together, dried with a gentle stream of nitrogen and reconstituted in 1 mL of MeOH/ACN (50:50, v/v) for the analysis. All analyses were done in triplicate.
2.5. Sample Collection
Sewage samples were collected from the sewage treatment plant of Las Palmas de Gran Canaria (Canary Islands, Spain). The samples were acidified to a pH between 2.5–3 and stored at 4 °C until analysis. Before analysis, samples were filtered using 0.45 µm filter.
3. Results and Discussion
3.1. Chemistry and Selectivity of Sol-Gel Poly (caprolactone-dimethylsiloxane-caprolactone) Coated FPSE Membrane
Commercially available sample preparation techniques, including solid phase microextraction (SPME), stir bar sorptive extraction (SBSE) and microextraction by packed sorbent (MEPS) often utilize pristine organic or inorganic polymers such as polydimethylsiloxane, poly(ethylene glycol), octadecylsilane as the sorbents. These pristine polymers possess their own intrinsic selectivity/affinity towards the target analytes and cannot be modified/improved to enhance the extraction efficiency. FPSE addresses the selectivity stalemate by uniquely incorporating three selectivity determinants in the device design: (a) a hydrophobic/hydrophilic fabric substrate; (b) an organic or inorganic polymer; and (c) a sol-gel precursor to chemically bind the polymer to the fabric substrate. The overall selectivity of the FPSE membrane depends on the nature of these three building blocks and can be easily modified or improved based on the analytical need. For example, application of tetramethyl orthosilicate in sol-gel silica coating transformed the coating into highly polar. Application of methyl trimethoxysilane in most of the sol-gel sorbents provided additional interaction via London dispersion. The utilization of 3-cyanopropyl trimethoxysilane in sol-gel CN-CW20M was to make the resulting sorbent more polar. All sol-gel sorbents were prepared using cotton cellulose fabric as the substrate. The hydrophilic nature of cotton cellulose fabric and its possession of abundant surface hydroxyl groups to anchor the sol-gel sorbent network with the substrate justify its selection as the substrate from a large number of substrate candidates.
Unlike commercial sample preparation techniques, the analytes extraction/preconcentration in FPSE is primarily carried out by different intermolecular interactions such as dipole–dipole interactions, London dispersion and hydrogen bonding between the analytes and the sol-gel based functional sorbents. Sol-gel derived sorbents are inherently porous with sponge-like surface morphology. As such, aqueous solution carrying the target analytes can easily permeate though the spongy sol-gel sorbent, interact rapidly via different intermolecular interactions and consequently get extracted. The built-in-porosity of the fabric substrate is retained even after the sol-gel sorbent coating. As a result, the aqueous sample can easily pass though the FPSE device that mimics a solid phase extraction disk. The through pores of the FPSE membrane also significantly contribute to accelerate the extraction kinetics. As can be seen in
Table 1, all three analytes are highly polar. It is logical to expect that polar sorbent such as sol-gel CN-CW20M, sol-gel CW20M, sol-gel PEG 300 would offer higher extraction efficiency compared to medium polar sorbent sol-gel poly (CAP-DMS-CAP). However, as the results demonstrate (
Table S1), sol-gel poly(CAP-DMS-CAP) was found as the optimum sorbent when both the sample volume and extraction time were taken into consideration. A closer look into the molecular make up of both the analytes (
Table 1) and the polymers (
Table 2) reveal that the analytes and poly(CAP-DMS-CAP) possess a large number of hydrogen donors and acceptors. In addition to other intermolecular interactions, hydrogen bonding interactions between the analytes and the FPSE membrane may have played an important role in the extraction mechanism, resulting in the high extraction efficiency of highly polar analytes on a medium polar sol-gel sorbent. The presented results again validate the necessity of sol-gel derived advanced sorbent systems possessing numerous and diversified functional groups.
3.2. Optimization of Fabric Phase Sorptive Extraction
The efficiency of sorption primarily depends upon the characteristics of the sorbent material used as well as the physicochemical properties of the analytes’ dissolution and the experimental conditions. Thus, the adsorption study was made using the seven fabric displays and an experimental design 2
3 with the variables that the most affect the adsorption. The variables and levels chosen were extraction time (15 and 30 min), sample volume (10 and 20 mL) and pH (3 and 9). The adsorption efficiency was calculated by the difference in the concentration of the spiked water before and after extraction. An aliquot of the sample is taken before the extraction and analyzed. After, the fabric is immersed in the sample during the extraction time and another aliquot of the water sample is taken for analysis. Results of all the fabrics tested are shown in the
Supplementary Material (Table S1).
As can be seen in the obtained results, the adsorption procedure in FPSE is strongly conditioned by the selected variables. The variation of one of the variables, although the other two remain constant, can lead to a considerable reduction in the adsorption capacity of the fabric. This condition makes it difficult to draw conclusions and predict the behavior.
In general, the best adsorption results were obtained at pH = 3, since in these conditions the analytes are in neutral form. However, for the fabrics CW20M and PEG 300 the better results were obtained at pH = 9. On the other hand, the fabric PTHF 250 was not greatly affected by this variable, since good adsorption results were obtained when the extraction time and the volume of sample were low, regardless of pH. For most of cases, an extraction time of 30 min obtained better results, but some fabrics, such as SICA and PCAP-DMS-CAP, obtained the best results with an extraction time of 15 min. Finally, in relation to the sample volume, most of the fabrics obtained a better adsorption efficiency when 10 mL was used. However, some fabrics, such as UCON and PCAP-DMS-CAP, achieved their best results when 20 mL was used.
Good adsorption efficiencies (>90%) were achieved in all fabrics tested under at least one of the conditions. Hence, for the selection of the fabric in the following tests, different factors were considered. A bigger sample volume would imply a higher preconcentration factor, so a fabric that obtained good results with 20 mL as sample volume was sought. On the other hand, a shorter extraction time implies a faster methodology, so a fabric that obtained the best results when extracted for 15 min was selected. With these considerations in mind, the fabric PCAP-DMS-CAP was selected for the next stage. Therefore, extraction conditions were set as follow: extraction time = 15 min., sample volume = 20 mL and pH = 3. In these conditions a mean adsorption (
n = 8) of 80 ± 9% of DOX, 85 ± 7% of DAU and 87 ± 7% of EPI on the selected fabric was obtained. All these results are presented in
Figure 2.
3.3. Optimization of Desorption Process
After adsorption conditions are defined, it is necessary to optimize the desorption conditions. To perform that, a 23 experimental design with the variables that most affect the elution process was done. The variables selected in this step were the elution time (3 and 7 min), the elution volume (0.5 and 1.5 mL) and the solvent type (MeOH and ACN). Recovery was measured by comparing the signal of a standard with the signal of the eluted.
After completing the whole design, a statistical study of the results was carried out through Pearson’s correlation and the
p-value. It was found that all the variables affect the process, as indicated by the
p-value; however, Pearson’s correlation did not indicate which value should take the variable (
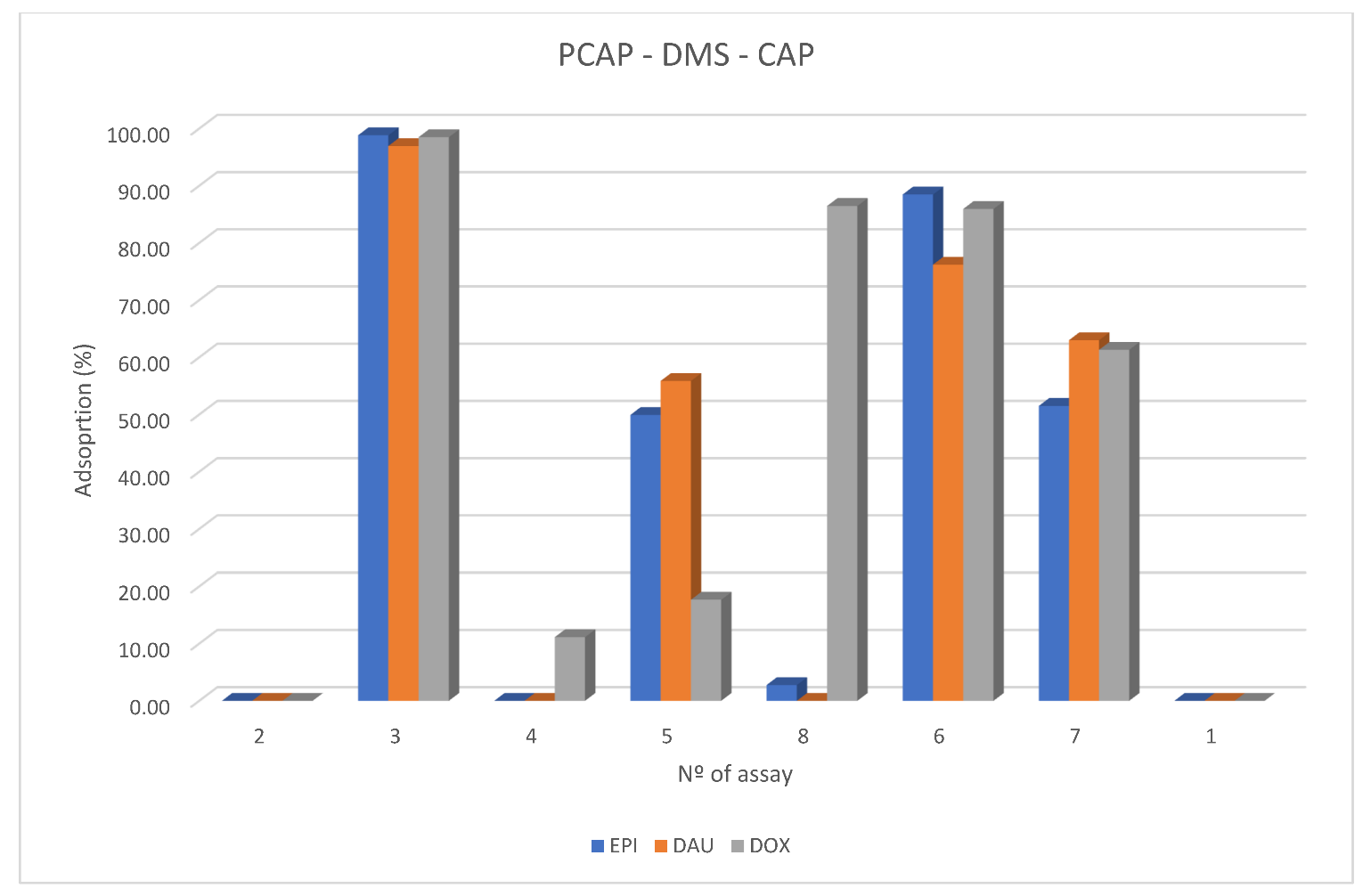
Table 3). Regarding to Pareto charts, it was only found for DOX that the pairwise combination of the variables was significant. EPI and DAU present a similar pattern, but their graphs were below the line of significance. For that reason, it was decided to select the intermediate values of each variable. In this sense, a new experimental design with 3
2 was prepared. The variables selected were solvent (MeOH, ACN and MeOH/ACN (50:50)) and elution time (4, 7 and 10 min.). A value of 1 mL was set for the elution volume due to that volume of 0.5 mL did not completely cover the fabric, which can lead to recovery and reproducibility errors. Results of this second experimental design are shown in
Figure 3. In the Figure, the solvents are codified as follows: MeOH = 1, MeOH/ACN (50:50) = 2 and ACN = 3.
As shown, the mixture of MeOH/ACN (50:50) achieved the best recovery values; however, the desorption of the analytes from the fabric is still not complete. FPSE is mainly an equilibrium extraction process, and the results show that, depending on the desorption time, the recovery of the analytes varies, possibly due to adsorption/desorption phenomena that take place until equilibrium is reached. More in depth, it was found that low desorption times benefit the elution, as can be seen in
Figure 2, for all of the compounds. Taking into account the results achieved so far, it was decided to set the desorption solvent as a mixture of 50:50 of MeOH/ACN, and to test lower desorption times. Thus, the elution time was set to 4 min. Minor times did not achieve better recoveries, probably because to reach equilibrium, more time is needed.
In order to increase the desorption efficiency, a new strategy was adopted to increase the solubility of the analytes in the solvent. The pH of the eluent was modified with ammonia or F.A. At first, 5% of ammonia and F.A. was tested, finding that a reduction in pH due to the addition of F.A. favored the recoveries and the increasing of the pH due to the ammonia did not improve the recovery; so the percentage of F.A. was increased until reaching the inflection point. The maximum desorption efficiency was found when 10% of F.A. was used. Then, with the selected eluent, a desorption in several steps was tried for even better elution. In it, the fabric was eluted with 1 mL of MeOH/ACN (50:50) + 10% F.A. for 4 min several times. It was found that the best desorption efficiency was achieved after carried out 2 elution steps. Then, the elutes were collected together, dried with nitrogen and reconstituted in 1 mL of MeOH/ACN (50:50) before injection.
In summary, the optimized procedure consists in immersing the selected fabric (PCAP-DMS-CAP) in 20 mL of the sample at pH = 3 for 15 min and stir at 1000 rpm. After that, the fabric is immersed in 1 mL of MeOH/ACN (50:50) + 10% F.A. for 4 min, twice. The eluted are collected together, dried with nitrogen and reconstituted in 1 mL of MeOH/ACN (50:50, v/v). Taking into account that the procedure started with 20 mL of the sample and it was preconcentrated up to 1 mL, a preconcentration factor of 20 was reached. This preconcentration factor was taken into account for the calculation of the analytical parameters.
3.4. Analytical Figures of Merit
To corroborate the linearity of the method, an external calibration curve was performed. In it, the relationship of peak area and the concentration of each compound was stablished. A linearity with a correlation coefficient (r2) higher than 0.990 was obtained for a concentration range from 25 to 500 µg·L−1 for all compounds.
To study the reproducibility and repeatability of the whole method FPSE-UHPLC-FD, intraday and interday studies were done at two level of concentrations (10 µg·L
−1 and 25 µg·L
−1, taking into account the preconcentration factor). The intraday relative standard deviation (RSD) was calculated by extracting 6 samples in the same day (
n = 6), while the interday RSD was calculated by extracting 3 samples (
n = 3) in 3 different days. Relative recovery was calculated taking into account the mean adsorption of the compounds into the fabric and in the same levels as before. It was calculated as the ratio between the extract of a spiked sample and a MeOH/ACN (50:50) standard. Limit of Detection (LOD) and Limit of Quantification (LOQ) were calculated as the signal to noise ratio higher than 3 and 10, respectively.
Table 4 show the analytical figures of merit obtained from the composite method.
It was found that the relative recoveries in Milli-Q water vary between 39 and 60%, depending on the compound, since it is an equilibrium process. Regarding the intraday and interday RSD, satisfactory values were found for all compounds at both levels. RSD values were lower than 15% for all the compounds in the two levels tested. Finally, LODs and LOQs suitable for the purpose of this work were achieved, ranging from 0.10–0.15 µg·L−1 and 0.33–0.49 µg·L−1, respectively. Comparing these results with those obtained before using microextraction techniques and FD, we have achieved a LOQ 2–3 times lower.
3.5. Application to Sewage Samples
To apply the developed methodology, sewage samples were analysed. However, the target compoundsof this study were not detected in any of the analyzed samples.
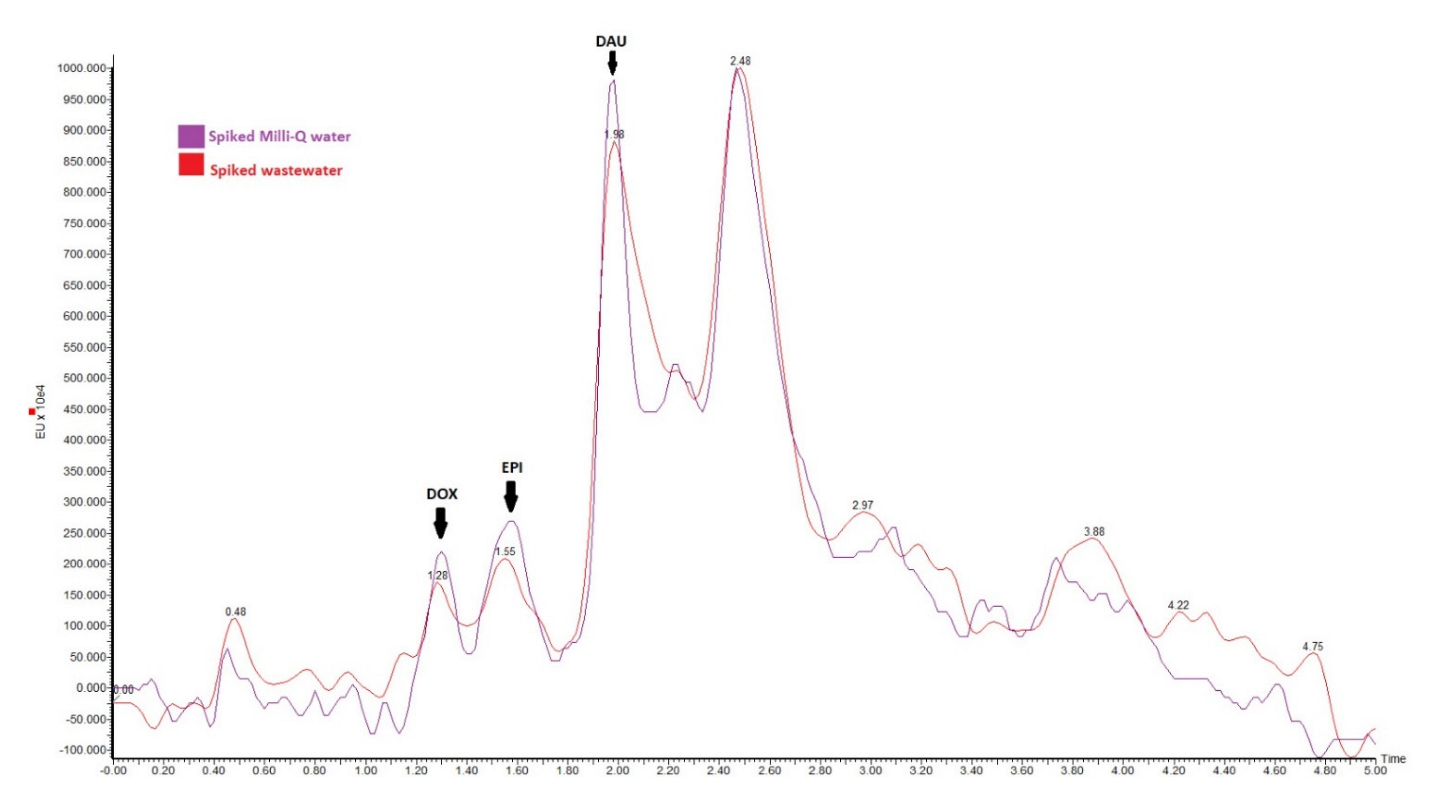
Therefore, in order to prove the applicability of the method, sewage samples were spiked at the same levels as the analytical parameters were done. It was found that the variation of the signal, depending on the compound and the concentration, varied from 80–110%, as can be seen in
Table 5 and
Figure 4. Unlike SPE extractions, which in many cases suffer significant signal suppression, the values obtained with this procedure indicate a great selectivity of the technique, as very low variations due to the matrix effect were obtained.
4. Conclusions
The available variety of sorbents for FPSE allows the selection of the most suitable for the mixture of analytes. Therefore, the optimization of a FPSE methodology leads to a low matrix effect, fast and easy extraction and low cost procedure.
This extraction technique was applied to the extraction and preconcentration of anthracyclines, a group of antineoplastic compounds used as chemotherapy treatment against different types, from sewage, prior to their determination by UHPLC-FD. In the optimal conditions, the extraction procedure obtains low RSD deviation, both intraday and interday, lower than 15% for all the compounds. Due to it being mainly an equilibrium process, mean recoveries ranging from 39% to 60% in Milli-Q water were obtained. Nevertheless, good LODs and LOQs were achieved, between 0.1–0.15 µg·L−1 and 0.33–0.49 µg·L−1, respectively. As a final point, this methodology was applied to sewage samples; however, analytes were not detected in the samples analyzed. Nevertheless, spiked samples prove the applicability of this fast method to a group of pharmaceuticals poorly studied, obtaining recoveries close to those obtained in Milli-Q water and without matrix effect, a common drawback of SPE and LC-MS/MS.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}


















