Molecular Alignment Under Strong Laser Pulses: Progress and Applications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Strong-Field Molecular Alignment: Progress
2.1. Dynamics of Molecular Alignment Under Linearly Polarized Laser Pulse
2.2. Advances in Techniques of Strong-Field Molecular Alignment
2.2.1. Pulse Sequence Techniques
2.2.2. Polarization Modulation Techniques
2.2.3. Pulse Shaping and Hybrid Methods
2.2.4. Theoretical Advances
3. Strong-Field Molecular Alignment: Applications
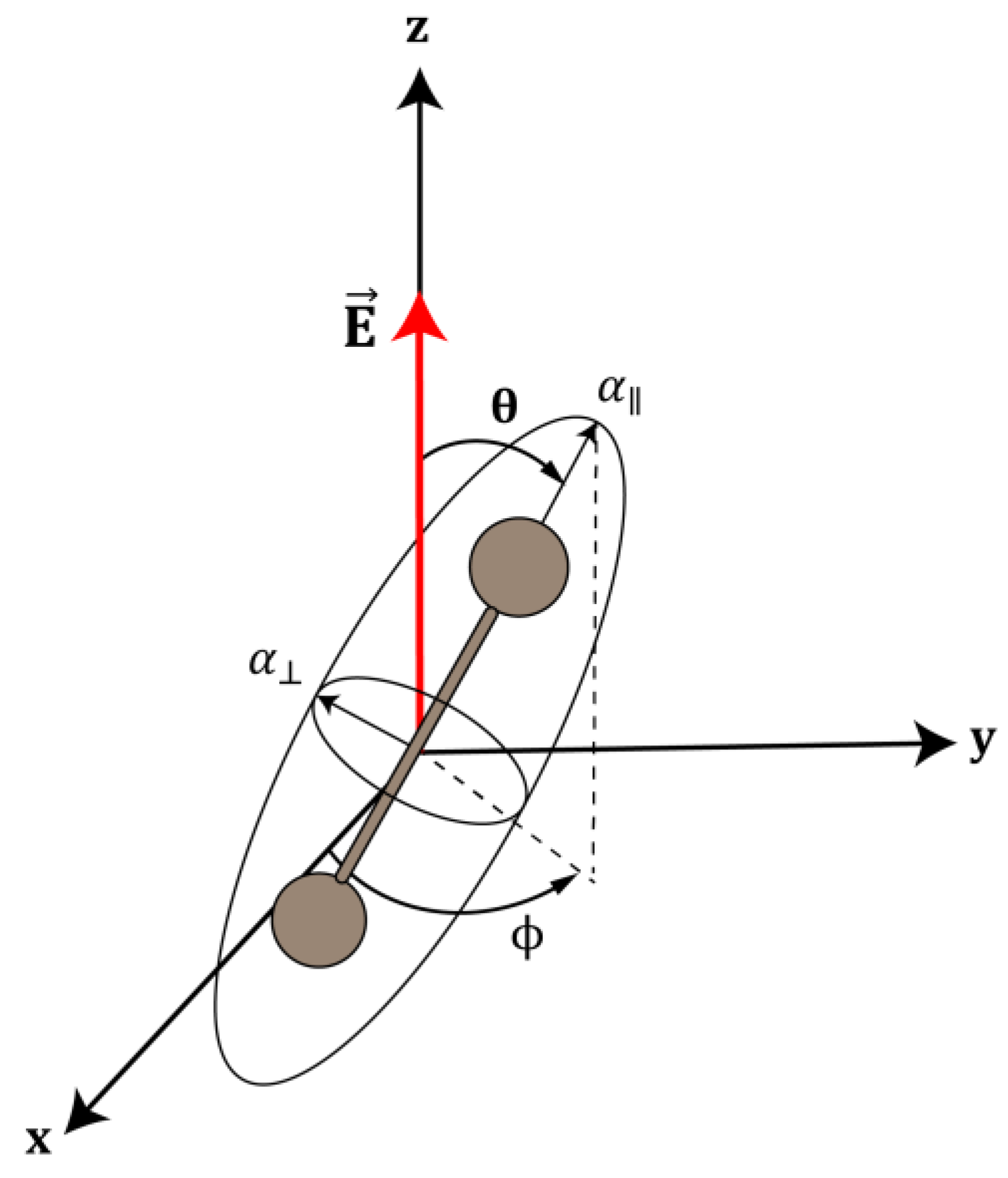
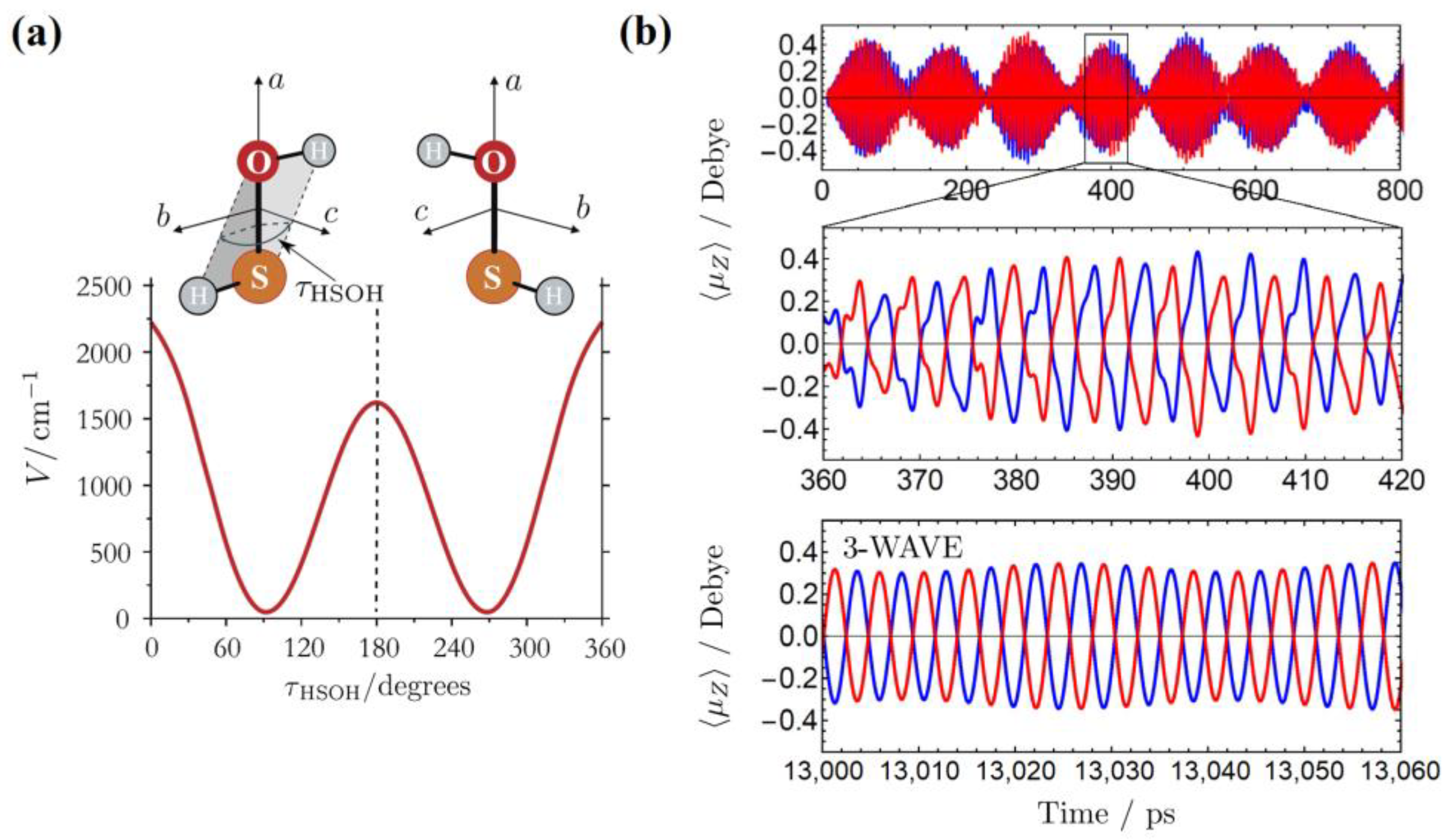
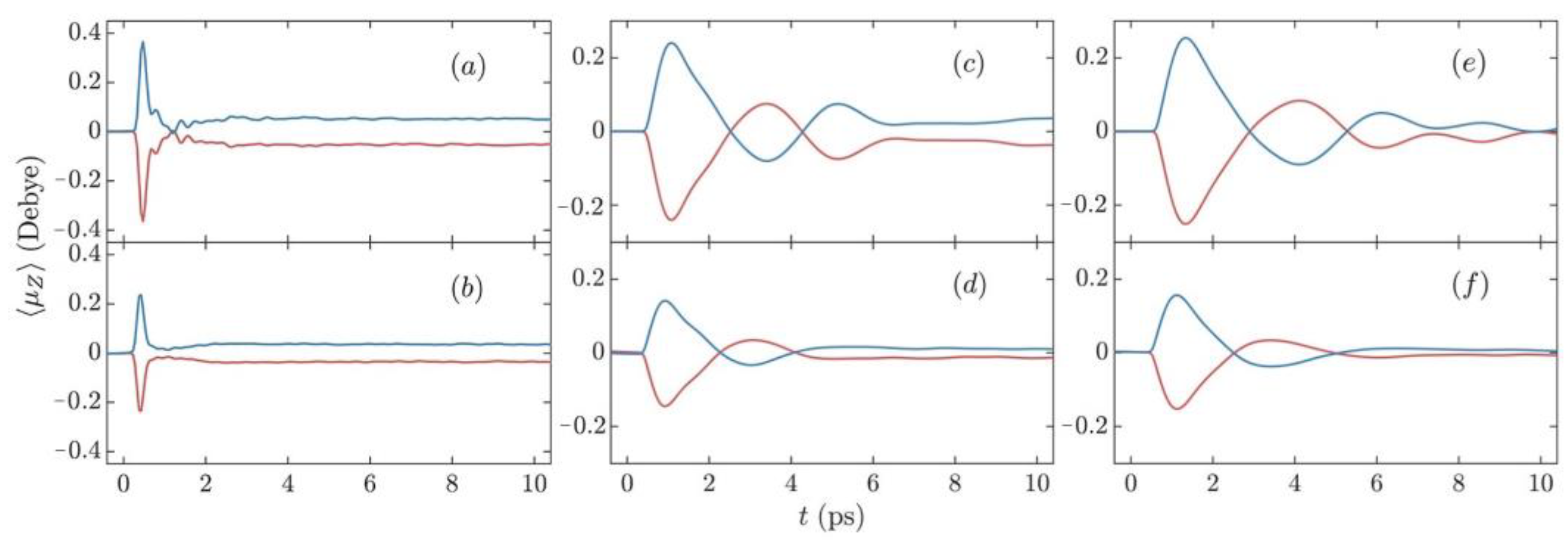
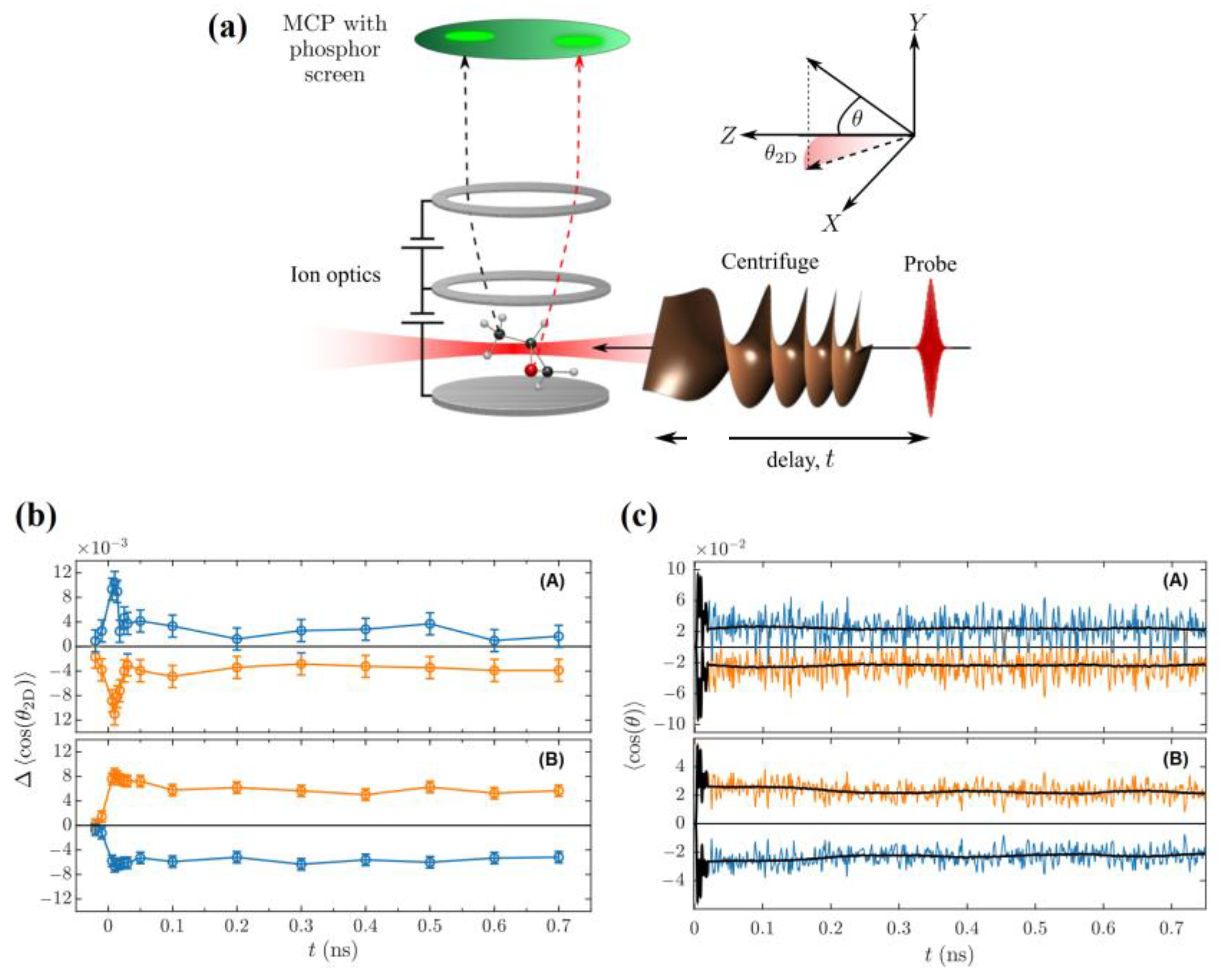
3.1. Enantioselective Orientation of Chiral Molecules
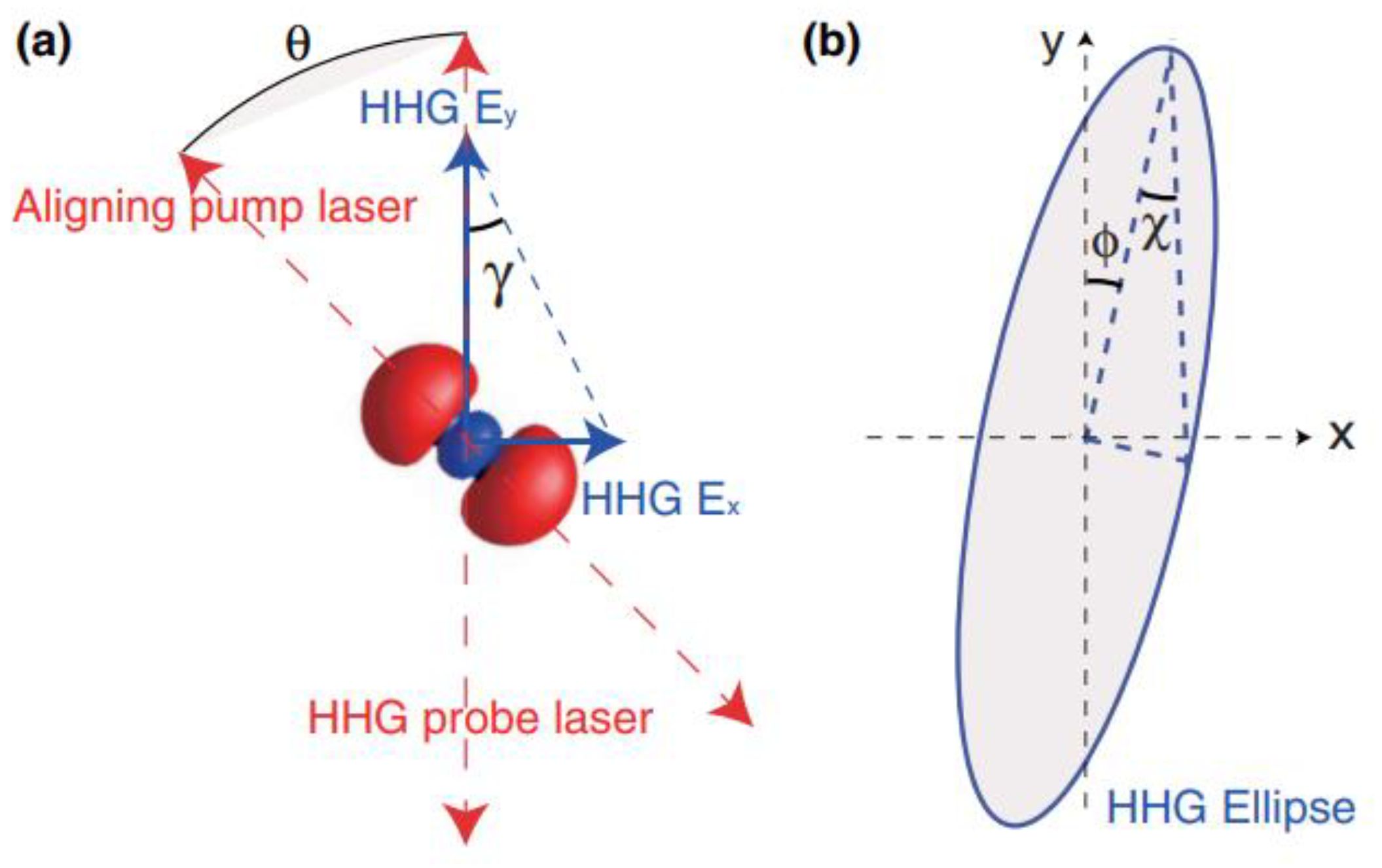
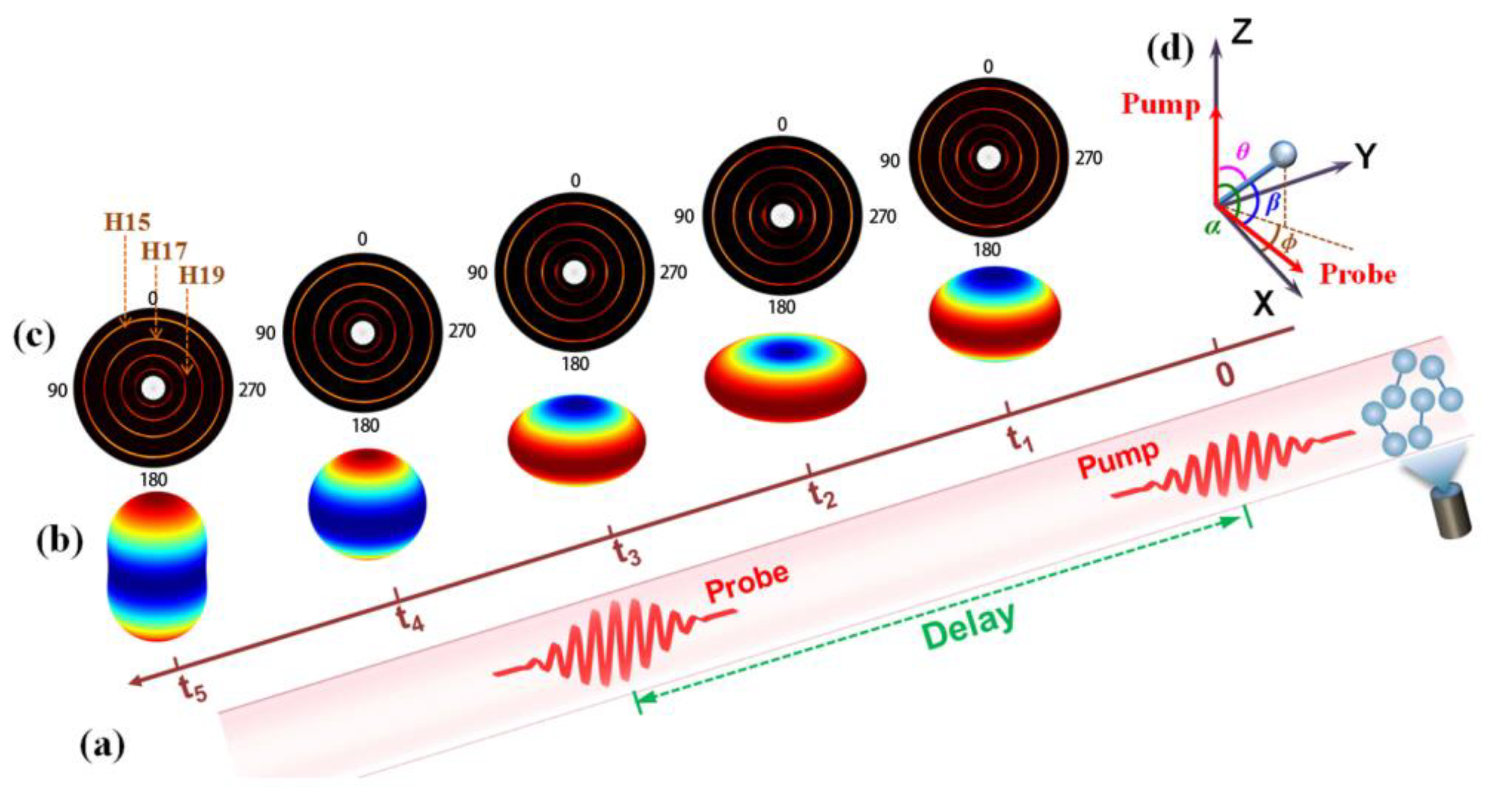
3.2. Alignment-Dependent HHG
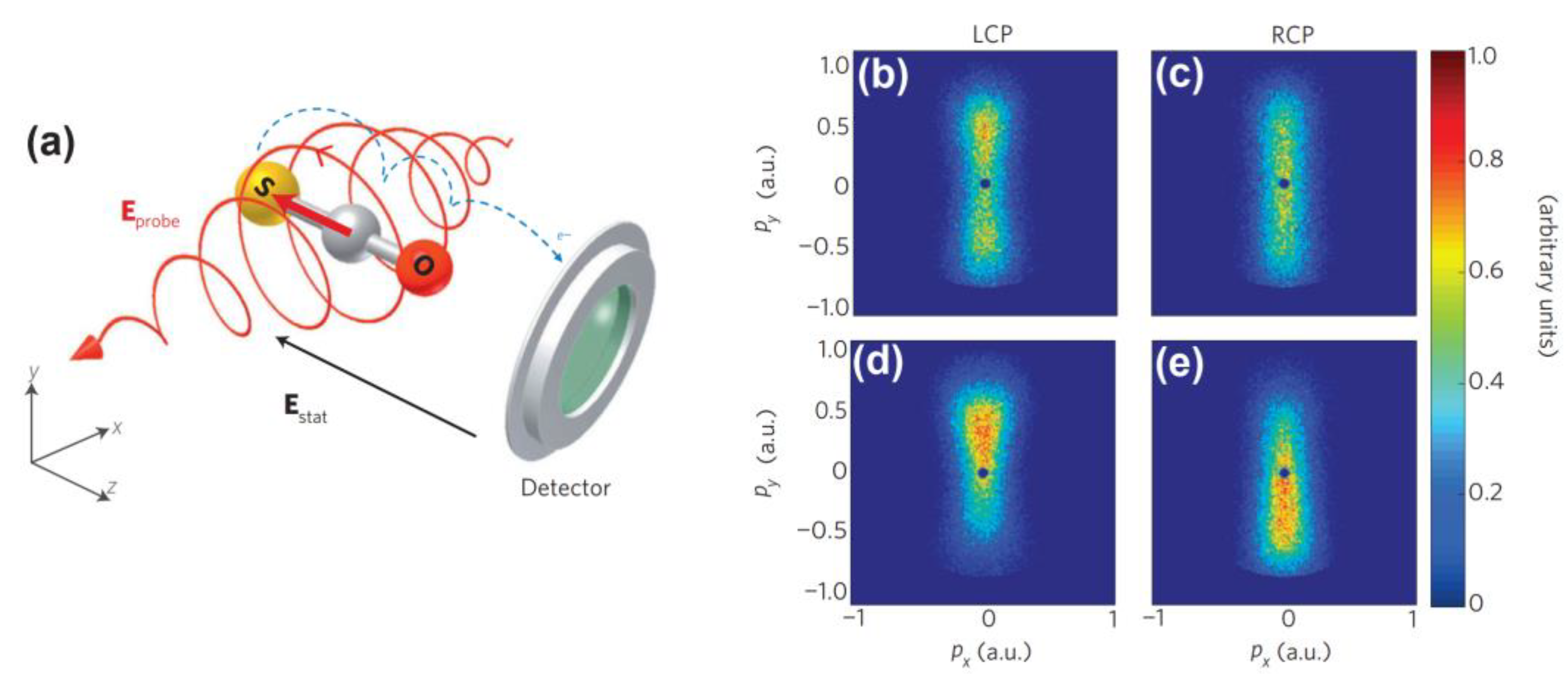
3.3. PADs and Ionization Efficiency via Molecular Alignment
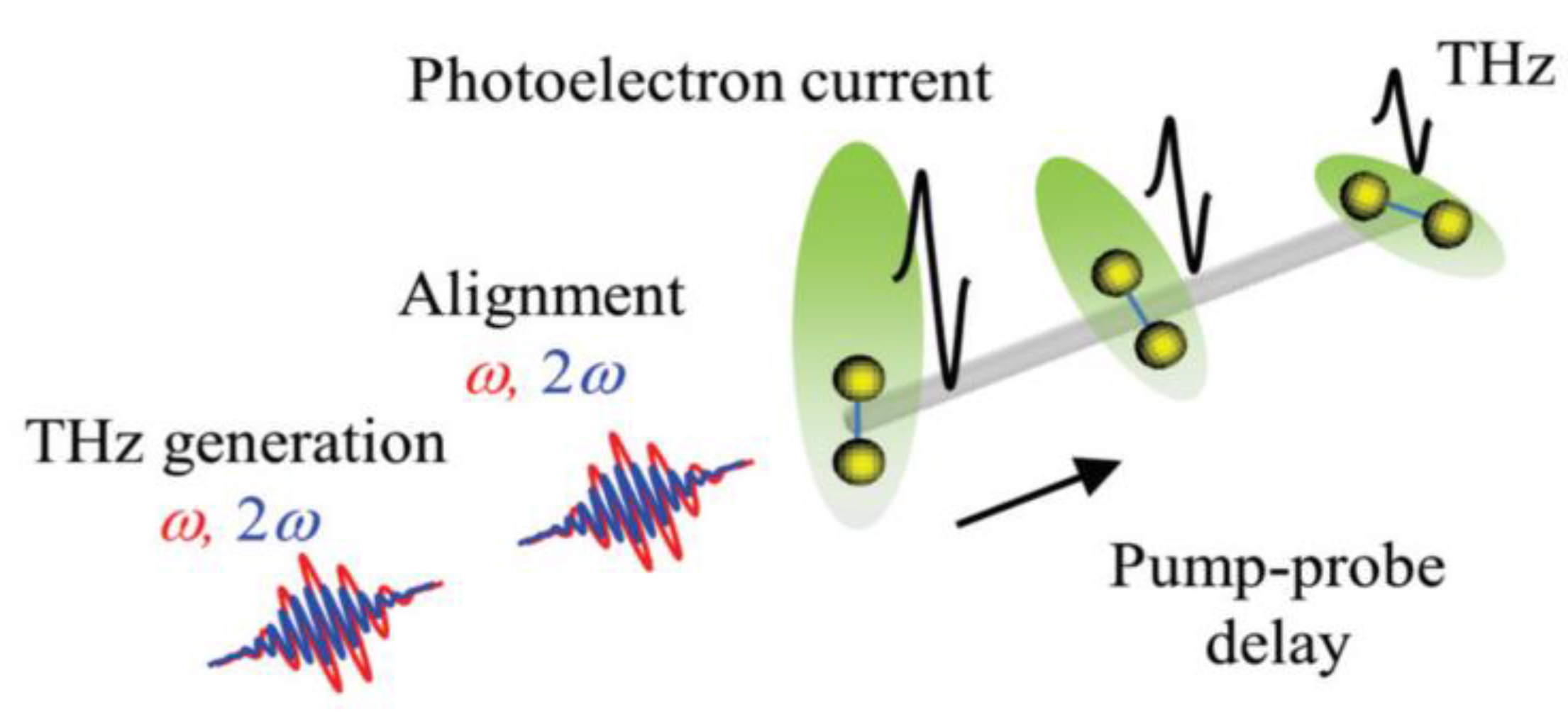
3.4. Alignment-Dependent THz Generation and Control
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Normand, D.; Lompre, L.; Cornaggia, C.; Physics, O. Laser-induced molecular alignment probed by a double-pulse experiment. J. Phys. B 1992, 25, L497. [Google Scholar] [CrossRef]
- Stapelfeldt, H.; Seideman, T. Colloquium: Aligning molecules with strong laser pulses. Rev. Mod. Phys. 2003, 75, 543–557. [Google Scholar] [CrossRef]
- Xu, N. One-Dimensional Molecular Alignment Induced by Femtosecond Laser Pulses. Ph.D. Thesis, Peking University, Beijing, China, 2008. [Google Scholar]
- Friedrich, B.; Herschbach, D. Alignment and trapping of molecules in intense laser fields. Phys. Rev. Lett. 1995, 74, 4623–4626. [Google Scholar] [CrossRef]
- Liu, F.C. Rotational State Selection and Alignment/Orientation of Molecules in External Fields. Ph.D. Thesis, Jilin University, Changchun, China, 2006. [Google Scholar]
- Spanner, M. Field-Free Alignment and Strong Field Control of Molecular Rotors. Ph.D. Thesis, University of Waterloo, Waterloo, ON, USA, 2004. [Google Scholar]
- Dooley, P.W.; Litvinyuk, I.V.; Lee, K.F.; Rayner, D.M.; Spanner, M.; Villeneuve, D.M.; Corkum, P.B. Direct imaging of rotational wave-packet dynamics of diatomic molecules. Phys. Rev. A 2003, 68, 023406. [Google Scholar] [CrossRef]
- De, S.; Znakovskaya, I.; Ray, D.; Anis, F.; Johnson, N.G.; Bocharova, I.A.; Magrakvelidze, M.; Esry, B.D.; Cocke, C.L.; Litvinyuk, I.V.; et al. Field-free orientation of CO molecules by femtosecond two-color laser fields. Phys. Rev. Lett. 2009, 103, 153002. [Google Scholar] [CrossRef] [PubMed]
- Renard, V.; Faucher, O.; Lavorel, B. Measurement of laser-induced alignment of molecules by cross defocusing. Opt. Lett. 2005, 30, 70–72. [Google Scholar] [CrossRef]
- Xu, N.; Wu, C.; Gao, Y.; Jiang, H.; Yang, H.; Gong, Q. Measurement of the field-free alignment of diatomic molecules. J. Phys. Chem. A 2008, 112, 612–617. [Google Scholar] [CrossRef]
- Fleischer, S.; Khodorkovsky, Y.; Prior, Y.; Sh Averbukh, I. Controlling the sense of molecular rotation. New J. Phys. 2009, 11, 105039. [Google Scholar] [CrossRef]
- Hoque, M.Z.; Lapert, M.; Hertz, E.; Billard, F.; Sugny, D.; Lavorel, B.; Faucher, O. Observation of laser-induced field-free permanent planar alignment of molecules. Phys. Rev. A 2011, 84, 013409. [Google Scholar] [CrossRef]
- Lin, K.; Song, Q.; Gong, X.; Ji, Q.; Pan, H.; Ding, J.; Zeng, H.; Wu, J. Visualizing molecular unidirectional rotation. Phys. Rev. A 2015, 92, 013410. [Google Scholar] [CrossRef]
- Lee, K.F.; Litvinyuk, I.V.; Dooley, P.W.; Spanner, M.; Villeneuve, D.M.; Corkum, P.B. Two-pulse alignment of molecules. J. Phys. B 2004, 37, L43–L48. [Google Scholar] [CrossRef]
- Bloomquist, C.; Zhdanovich, S.; Milner, A.A.; Milner, V. Directional spinning of molecules with sequences of femtosecond pulses. Phys. Rev. A 2012, 86, 063413. [Google Scholar] [CrossRef]
- Fleischer, S.; Averbukh, I.S.; Prior, Y. Selective control of molecular rotation. J. Phys. B 2008, 41, 074018. [Google Scholar] [CrossRef]
- Zhdanovich, S.; Milner, A.A.; Bloomquist, C.; Floss, J.; Averbukh, I.; Hepburn, J.W.; Milner, V. Control of molecular rotation with a chiral train of ultrashort pulses. Phys. Rev. Lett. 2011, 107, 243004. [Google Scholar] [CrossRef]
- Cryan, J.P.; Bucksbaum, P.H.; Coffee, R.N. Field-free alignment in repetitively kicked nitrogen gas. Phys. Rev. A 2009, 80, 063412. [Google Scholar] [CrossRef]
- Larsen, J.J.; Sakai, H.; Safvan, C.P.; Wendt-Larsen, I.; Stapelfeldt, H. Aligning molecules with intense nonresonant laser fields. J. Chem. Phys. 1999, 111, 7774–7781. [Google Scholar] [CrossRef]
- Larsen, J.J.; Hald, K.; Bjerre, N.; Stapelfeldt, H.; Seideman, T. Three dimensional alignment of molecules using elliptically polarized laser fields. Phys. Rev. Lett. 2000, 85, 2470–2473. [Google Scholar] [CrossRef]
- Jingsong Liu, J.L.; Qiyuan Cheng, Q.C.; Daguang Yue, D.Y.; Xucong Zhou, X.Z.; Qingtian Meng, Q.M. Dynamical analysis of the effect of elliptically polarized laser pulses on molecular alignment and orientation. Chin. Opt. Lett. 2018, 16, 103201. [Google Scholar] [CrossRef]
- Lapert, M.; Hertz, E.; Guérin, S.; Sugny, D. Field-free permanent molecular planar alignment. Phys. Rev. A 2009, 80, 051403. [Google Scholar] [CrossRef]
- Lee, K.F.; Villeneuve, D.M.; Corkum, P.B.; Stolow, A.; Underwood, J.G. Field-free three-dimensional alignment of polyatomic molecules. Phys. Rev. Lett. 2006, 97, 173001. [Google Scholar] [CrossRef]
- Underwood, J.G.; Sussman, B.J.; Stolow, A. Field-free three dimensional molecular axis alignment. Phys. Rev. Lett. 2005, 94, 143002. [Google Scholar] [CrossRef] [PubMed]
- Chatterley, A.S.; Shepperson, B.; Stapelfeldt, H. Three-Dimensional Molecular Alignment Inside Helium Nanodroplets. Phys. Rev. Lett. 2017, 119, 073202. [Google Scholar] [CrossRef]
- Lin, K.; Tutunnikov, I.; Qiang, J.; Ma, J.; Song, Q.; Ji, Q.; Zhang, W.; Li, H.; Sun, F.; Gong, X. All-optical field-free three-dimensional orientation of asymmetric-top molecules. Nat. Commun. 2018, 9, 5134. [Google Scholar] [CrossRef] [PubMed]
- Machholm, M.; Henriksen, N.E. Field-free orientation of molecules. Phys. Rev. Lett. 2001, 87, 193001. [Google Scholar] [CrossRef]
- Fleischer, S.; Zhou, Y.; Field, R.W.; Nelson, K.A. Molecular orientation and alignment by intense single-cycle THz pulses. Phys. Rev. Lett. 2011, 107, 163603. [Google Scholar] [CrossRef]
- Xu, S.; Yao, Y.; Lu, C.; Ding, J.; Jia, T.; Zhang, S.; Sun, Z. Manipulating field-free molecular alignment byV-shaped femtosecond laser pulses. Phys. Rev. A 2014, 89, 053420. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, S.; Zhang, S. Selective excitation and control of the molecular orientation by a phase shaped laser pulse. RSC Adv. 2016, 6, 100295–100299. [Google Scholar] [CrossRef]
- Karczmarek, J.; Wright, J.; Corkum, P.; Ivanov, M. Optical Centrifuge for Molecules. Phys. Rev. Lett. 1999, 82, 3420–3423. [Google Scholar] [CrossRef]
- Milner, A.A.; Korobenko, A.; Milner, V. Field-free long-lived alignment of molecules with a two-dimensional optical centrifuge. Phys. Rev. A 2016, 93, 053408. [Google Scholar] [CrossRef]
- Takei, D.; Mun, J.H.; Minemoto, S.; Sakai, H. Laser-field-free three-dimensional molecular orientation. Phys. Rev. A 2016, 94, 013401. [Google Scholar] [CrossRef]
- Mullins, T.; Karamatskos, E.T.; Wiese, J.; Onvlee, J.; Rouzee, A.; Yachmenev, A.; Trippel, S.; Kupper, J. Picosecond pulse-shaping for strong three-dimensional field-free alignment of generic asymmetric-top molecules. Nat. Commun. 2022, 13, 1431. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.-C.; Henriksen, N.E. Field-free molecular orientation induced by single-cycle THz pulses: The role of resonance and quantum interference. Phys. Rev. A 2013, 87, 013408. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Yu, J.; Cong, S.-L. Field-free molecular orientation by two-color shaped laser pulse together with time-delayed THz laser pulse. Laser Phys. Lett. 2013, 10, 076001. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, H.; Lavorel, B.; Billard, F.; Hertz, E.; Wu, J.; Boulet, C.; Hartmann, J.-M.; Faucher, O. Observing Collisions beyond the Secular Approximation Limit. Nat. Commun. 2019, 10, 5780. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xu, L.; Zhou, L.; Shi, M.; Lu, P.; Lin, K.; Wu, J. Laser-Induced k-Alignment Echoes and 4pi Phase-Space Analysis. Phys. Rev. A 2024, 110, 33102. [Google Scholar] [CrossRef]
- Karras, G.; Hertz, E.; Billard, F.; Lavorel, B.; Hartmann, J.-M.; Faucher, O.; Gershnabel, E.; Prior, Y.; Averbukh, I.S. Orientation and Alignment Echoes. Phys. Rev. Lett. 2015, 114, 153601. [Google Scholar] [CrossRef]
- Lin, K.; Lu, P.; Ma, J.; Gong, X.; Song, Q.; Ji, Q.; Zhang, W.; Zeng, H.; Wu, J.; Karras, G.; et al. Echoes in Space and Time. Phys. Rev. X 2016, 6, 41056. [Google Scholar] [CrossRef]
- Zhang, H.; Lavorel, B.; Billard, F.; Hartmann, J.-M.; Hertz, E.; Faucher, O.; Ma, J.; Wu, J.; Gershnabel, E.; Prior, Y.; et al. Rotational Echoes as a Tool for Investigating Ultrafast Collisional Dynamics of Molecules. Phys. Rev. Lett. 2019, 122, 193401. [Google Scholar] [CrossRef]
- Li, S.-Y.; Guo, F.-M.; Wang, J.; Yang, Y.-J.; Jin, M.-X. Field-free orientation of diatomic molecule via the linearly polarized resonant pulses. Chin. Phys. B 2015, 24, 104205. [Google Scholar] [CrossRef]
- Tyagi, A.; Maan, A.; Ahlawat, D.S.; Prasad, V. Effect of aligning pulse train on the orientation and alignment of a molecule in presence of orienting pulse. Spectrochim. Acta A 2017, 173, 13–18. [Google Scholar] [CrossRef]
- Kitano, K.; Hasegawa, H.; Ohshima, Y. Ultrafast angular momentum orientation by linearly polarized laser fields. Phys. Rev. Lett. 2009, 103, 223002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Wang, S.; Henriksen, N.E. Optimal field-free orientation of molecules with resonant tailored multicolor fields. Phys. Rev. A 2023, 107, 033118. [Google Scholar] [CrossRef]
- Dang, H.-P.; Wang, S.; Zhan, W.-S.; Zai, J.-B.; Han, X. Field-free orientation dynamics of CO molecule by combining two-color shaped laser pulse with THz laser pulse train. Chem. Phys. 2015, 461, 81–88. [Google Scholar] [CrossRef]
- Kraus, P.M.; Baykusheva, D.; Worner, H.J. Two-pulse field-free orientation reveals anisotropy of molecular shape resonance. Phys. Rev. Lett. 2014, 113, 023001. [Google Scholar] [CrossRef]
- Egodapitiya, K.N.; Li, S.; Jones, R.R. Terahertz-induced field-free orientation of rotationally excited molecules. Phys. Rev. Lett. 2014, 112, 103002. [Google Scholar] [CrossRef] [PubMed]
- Trippel, S.; Mullins, T.; Muller, N.L.; Kienitz, J.S.; Gonzalez-Ferez, R.; Kupper, J. Two-state wave packet for strong field-free molecular orientation. Phys. Rev. Lett. 2015, 114, 103003. [Google Scholar] [CrossRef]
- Chi, F.; Yang, Y.; Guo, F. Controlling field-free molecular orientation with combined circularly and linearly polarized resonant pulses at low rotational temperatures. Chem. Phys. Lett. 2013, 556, 350–354. [Google Scholar] [CrossRef]
- Sonoda, K.; Iwasaki, A.; Yamanouchi, K.; Hasegawa, H. Field-free molecular orientation of nonadiabatically aligned OCS. Chem. Phys. Lett. 2018, 693, 114–120. [Google Scholar] [CrossRef]
- Yachmenev, A.; Yurchenko, S.N. Detecting Chirality in Molecules by Linearly Polarized Laser Fields. Phys. Rev. Lett. 2016, 117, 033001. [Google Scholar] [CrossRef]
- Patterson, D.; Doyle, J.M. Sensitive chiral analysis via microwave three-wave mixing. Phys. Rev. Lett. 2013, 111, 023008. [Google Scholar] [CrossRef]
- Gershnabel, E.; Averbukh, I.S. Orienting Asymmetric Molecules by Laser Fields with Twisted Polarization. Phys. Rev. Lett. 2018, 120, 083204. [Google Scholar] [CrossRef]
- Tutunnikov, I.; Gershnabel, E.; Gold, S.; Averbukh, I.S. Selective Orientation of Chiral Molecules by Laser Fields with Twisted Polarization. J. Phys. Chem. Lett. 2018, 9, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Tutunnikov, I.; Floß, J.; Gershnabel, E.; Brumer, P.; Averbukh, I.S.; Milner, A.A.; Milner, V. Observation of persistent orientation of chiral molecules by a laser field with twisted polarization. Phys. Rev. A 2020, 101, 021403. [Google Scholar] [CrossRef]
- Milner, A.A.; Fordyce, J.A.M.; MacPhail-Bartley, I.; Wasserman, W.; Milner, V.; Tutunnikov, I.; Averbukh, I.S. Controlled Enantioselective Orientation of Chiral Molecules with an Optical Centrifuge. Phys. Rev. Lett. 2019, 122, 223201. [Google Scholar] [CrossRef] [PubMed]
- Tessari, Z.; Tahara, K.; De Feyter, S. Molecular Alignment and Chiral Bias under Dynamic Nanoconfinement Conditions: The Impact of Molecular Symmetry. J. Phys. Chem. C 2023, 127, 9248–9256. [Google Scholar] [CrossRef]
- Velotta, R.; Hay, N.; Mason, M.B.; Castillejo, M.; Marangos, J.P. High-Order Harmonic Generation in Aligned Molecules. Phys. Rev. Lett. 2001, 87, 183901. [Google Scholar] [CrossRef]
- McFarland, B.K.; Farrell, J.P.; Bucksbaum, P.H.; Guhr, M. High harmonic generation from multiple orbitals in N2. Science 2008, 322, 1232–1235. [Google Scholar] [CrossRef]
- Zhou, X.; Lock, R.; Wagner, N.; Li, W.; Kapteyn, H.C.; Murnane, M.M. Elliptically polarized high-order harmonic emission from molecules in linearly polarized laser fields. Phys. Rev. Lett. 2009, 102, 073902. [Google Scholar] [CrossRef]
- Le, A.-T.; Lucchese, R.R.; Lin, C.D. Polarization and ellipticity of high-order harmonics from aligned molecules generated by linearly polarized intense laser pulses. Phys. Rev. A 2010, 82, 023814. [Google Scholar] [CrossRef]
- He, Y.; He, L.; Lan, P.; Wang, B.; Li, L.; Zhu, X.; Cao, W.; Lu, P. Direct imaging of molecular rotation with high-order-harmonic generation. Phys. Rev. A 2019, 99, 053419. [Google Scholar] [CrossRef]
- Jiang, C.; Jiang, H.; Chen, Y.; Li, B.; Lin, C.; Jin, C. Genetic-algorithm retrieval of the molecular alignment distribution with high-order harmonics generated from transiently aligned CO2 molecules. Phys. Rev. A 2022, 105, 023111. [Google Scholar] [CrossRef]
- Li, H.; Franks, K.J.; Hanson, R.J.; Kong, W. Brute Force Orientation and Alignment of Pyridazine Probed by Resonantly Enhanced Multiphoton Ionization. J. Phys. Chem. A 1998, 102, 8084–8090. [Google Scholar] [CrossRef]
- Holmegaard, L.; Hansen, J.L.; Kalhøj, L.; Louise Kragh, S.; Stapelfeldt, H.; Filsinger, F.; Küpper, J.; Meijer, G.; Dimitrovski, D.; Abu-samha, M.; et al. Photoelectron angular distributions from strong-field ionization of oriented molecules. Nat. Phys. 2010, 6, 428–432. [Google Scholar] [CrossRef]
- Jin, C.; Le, A.-T.; Zhao, S.-F.; Lucchese, R.; Lin, C.J.P.R.A. Theoretical study of photoelectron angular distributions in single-photon ionization of aligned N2 and CO2. Phys. Rev. A 2010, 81, 033421. [Google Scholar] [CrossRef]
- Thomann, I.; Lock, R.; Sharma, V.; Gagnon, E.; Pratt, S.T.; Kapteyn, H.C.; Murnane, M.M.; Li, W. Direct measurement of the angular dependence of the single-photon ionization of aligned N2 and CO2. J. Phys. Chem. A 2008, 112, 9382–9386. [Google Scholar] [CrossRef]
- Liu, X.; Gao, S.; Wang, N.; Zhu, F.; Jiao, L.; Liu, A. Alignment sensitivity of molecular-frame photoelectron momentum and angular distribution of H2+by XUV attosecond pulses. Commun. Theor. Phys. 2023, 75, 035503. [Google Scholar] [CrossRef]
- Tsubouchi, M.; Whitaker, B.J.; Wang, L.; Kohguchi, H.; Suzuki, T. Photoelectron imaging on time-dependent molecular alignment created by a femtosecond laser pulse. Phys. Rev. Lett. 2001, 86, 4500–4503. [Google Scholar] [CrossRef]
- Zhou, M.; Guo, M.Q.; Yu, J.; Chai, S.; Cong, S.L. The influence of molecular alignment and orientation in the ground state and excited state on the resonance-enhanced multi-photon ionization dynamics. Spectrochim. Acta A 2021, 262, 120154. [Google Scholar] [CrossRef]
- You, Y.; Oh, T.; Fallahkhair, A.; Kim, K. Alignment-dependent terahertz radiation in two-color photoionization of molecules. Phys. Rev. A 2013, 87, 035401. [Google Scholar] [CrossRef]
- Chen, W.; Huang, Y.; Meng, C.; Liu, J.; Zhou, Z.; Zhang, D.; Yuan, J.; Zhao, Z. Theoretical study of terahertz generation from atoms and aligned molecules driven by two-color laser fields. Phys. Rev. A 2015, 92, 033410. [Google Scholar] [CrossRef]
- Wu, J.; Tong, Y.; Li, M.; Pan, H.; Zeng, H. THz generation by a two-color pulse in prealigned molecules. Phys. Rev. A 2010, 82, 053416. [Google Scholar] [CrossRef]
- Li, M.; Pan, H.; Tong, Y.; Chen, C.; Shi, Y.; Wu, J.; Zeng, H. All-optical ultrafast polarization switching of terahertz radiation by impulsive molecular alignment. Opt. Lett. 2011, 36, 3633–3635. [Google Scholar] [CrossRef]
- Béjot, P.; Petit, Y.; Bonacina, L.; Kasparian, J.; Moret, M.; Wolf, J.-P. Ultrafast gaseous “half-wave plate”. Opt. Express 2008, 16, 7564–7570. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Meng, C.; Wang, X.; Lu, Z.; Zhang, D.; Chen, W.; Zhao, J.; Yuan, J.; Zhao, Z. Joint Measurements of Terahertz Wave Generation and High-Harmonic Generation from Aligned Nitrogen Molecules Reveal Angle-Resolved Molecular Structures. Phys. Rev. Lett. 2015, 115, 123002. [Google Scholar] [CrossRef]
- Li, Y.-N.; Du, J.-X.; Zhang, L.; Jiao, Z.-H.; Zhao, S.-F.; Wang, G.-L. Retrieval of the molecular alignment distribution from THz generation. Phys. Rev. A 2023, 108, 063103. [Google Scholar] [CrossRef]
- Xu, L.; Tutunnikov, I.; Prior, Y.; Averbukh, I.S. Enhanced persistent orientation of asymmetric-top molecules induced by cross-polarized terahertz pulses. Phys. Rev. Res. 2024, 6, 013201. [Google Scholar] [CrossRef]
- Rohrbach, D.; Beéry, L.; Mousavi, S.J.; Frey, H.M.; Kang, B.J.; Zyaee, E.; Feurer, T. THz-induced gas alignment in dispersionless field enhancing waveguides. Opt. Express 2025, 33, 3315–3324. [Google Scholar] [CrossRef]
- Cercignani, C. The Boltzmann Equation; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Kunitski, M.; Guan, Q.; Maschkiwitz, H.; Hahnenbruch, J.; Eckart, S.; Zeller, S.; Kalinin, A.; Schöffler, M.; Schmidt, L.P.H.; Jahnke, T.; et al. Ultrafast Manipulation of the Weakly Bound Helium Dimer. Nat. Phys. 2021, 17, 174–178. [Google Scholar] [CrossRef]
- Wu, J.; Vredenborg, A.; Ulrich, B.; Schmidt, L.P.H.; Meckel, M.; Voss, S.; Sann, H.; Kim, H.; Jahnke, T.; Dörner, R. Nonadiabatic Alignment of van Der Waals--Force-Bound Argon Dimers by Femtosecond Laser Pulses. Phys. Rev. A 2011, 83, 61403. [Google Scholar] [CrossRef]
- Lu, C.; Xu, L.; Zhou, L.; Shi, M.; Lu, P.; Li, W.; Dörner, R.; Lin, K.; Wu, J. Intermolecular Interactions Probed by Rotational Dynamics in Gas-Phase Clusters. Nat. Commun. 2024, 15, 4360. [Google Scholar] [CrossRef]
- Von Veltheim, A.; Borchers, B.; Steinmeyer, G.; Rottke, H. Imaging the Impulsive Alignment of Noble-Gas Dimers via Coulomb Explosion. Phys. Rev. A 2014, 89, 23432. [Google Scholar] [CrossRef]
- Pentlehner, D.; Nielsen, J.H.; Slenczka, A.; Mølmer, K.; Stapelfeldt, H. Impulsive Laser Induced Alignment of Molecules Dissolved in Helium Nanodroplets. Phys. Rev. Lett. 2013, 110, 93002. [Google Scholar] [CrossRef]
- Chatterley, A.S.; Schouder, C.; Christiansen, L.; Shepperson, B.; Rasmussen, M.H.; Stapelfeldt, H. Long-Lasting Field-Free Alignment of Large Molecules inside Helium Nanodroplets. Nat. Commun. 2019, 10, 133. [Google Scholar] [CrossRef]
- Chatterley, A.S.; Christiansen, L.; Schouder, C.A.; Jørgensen, A.V.; Shepperson, B.; Cherepanov, I.N.; Bighin, G.; Zillich, R.E.; Lemeshko, M.; Stapelfeldt, H. Rotational Coherence Spectroscopy of Molecules in Helium Nanodroplets: Reconciling the Time and the Frequency Domains. Phys. Rev. Lett. 2020, 125, 13001. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, I.N.; Bighin, G.; Schouder, C.A.; Chatterley, A.S.; Albrechtsen, S.H.; Muñoz, A.V.; Christiansen, L.; Stapelfeldt, H.; Lemeshko, M. Excited Rotational States of Molecules in a Superfluid. Phys. Rev. A 2021, 104, L061303. [Google Scholar] [CrossRef]
- Qiang, J.; Zhou, L.; Lu, P.; Lin, K.; Ma, Y.; Pan, S.; Lu, C.; Jiang, W.; Sun, F.; Zhang, W.; et al. Femtosecond Rotational Dynamics of D2 Molecules in Superfluid Helium Nanodroplets. Phys. Rev. Lett. 2022, 128, 243201. [Google Scholar] [CrossRef] [PubMed]
- Shepperson, B.; Søndergaard, A.A.; Christiansen, L.; Kaczmarczyk, J.; Zillich, R.E.; Lemeshko, M.; Stapelfeldt, H. Laser-Induced Rotation of Iodine Molecules in Helium Nanodroplets: Revivals and Breaking Free. Phys. Rev. Lett. 2017, 118, 203203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.; Guo, D.; Zhao, L.; Yu, L.; Zhang, H.; He, Y.J. Great concern for chiral pharmaceuticals from the thalidomide tragedy. Univ. Chem. 2019, 34, 1–12. [Google Scholar] [CrossRef]
- Corkum, P.B. Plasma perspective on strong-field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhang, E.; Liang, Q.; Liu, Y. Molecular Alignment Under Strong Laser Pulses: Progress and Applications. Photonics 2025, 12, 422. https://doi.org/10.3390/photonics12050422
Wang M, Zhang E, Liang Q, Liu Y. Molecular Alignment Under Strong Laser Pulses: Progress and Applications. Photonics. 2025; 12(5):422. https://doi.org/10.3390/photonics12050422
Chicago/Turabian StyleWang, Ming, Enliang Zhang, Qingqing Liang, and Yi Liu. 2025. "Molecular Alignment Under Strong Laser Pulses: Progress and Applications" Photonics 12, no. 5: 422. https://doi.org/10.3390/photonics12050422
APA StyleWang, M., Zhang, E., Liang, Q., & Liu, Y. (2025). Molecular Alignment Under Strong Laser Pulses: Progress and Applications. Photonics, 12(5), 422. https://doi.org/10.3390/photonics12050422