1. Introduction
The effects of metallic surfaces and nanostructures on the optical properties (fluorescence quenching or enhancement) of molecules nearby were discussed already in the late 1960s and 1970s [
1,
2,
3]. The fluorescence enhancement and photostability of those molecules due to plasmonic nanostructures in their vicinity influenced many research areas. Based on altered spontaneous emission rates, various devices, e.g., surface plasmon-enhanced LEDs [
4], plasmon-enhanced optical sensors [
5,
6], single-photon sources for quantum cryptography [
7] and photonic crystal lasers [
8,
9], have been revealed. In 2002, Geddes
et al. considered the outcome of the near-field of metallic nanoparticles on the brightness and photostability of chromophores for the first time and introduced the term metal-enhanced fluorescence (MEF) [
10]. Details of the real mechanism of MEF are still part of the discussion today. However, changes in the local field stimulated by metallic nanostructures and modifications in radiative decay rates are two acknowledged procedures for the fluorescence enhancement [
8,
11,
12]. The fluorescence of fluorophores can be controlled by controlling the distance between fluorophore and plasmonic active particles, their relative orientation, size, shape and the surrounding medium [
11]. The interaction of plasmonic nanostructures with fluorophores increases their radiative decay rates and quantum yield. The increased decay rates yield an increase of the photostability of fluorophores, as the fluorophores spend less time in an excited state before returning back to the ground state [
13]. The ability to increase the brightness of the fluorophore with adjacent plasmonic nanostructures simplifies the detection of the fluorophores at the single-molecule level, which otherwise is difficult due to low signal intensities.
The prior studies of our group showed the influence of plasmonic interactions on the fluorescence intensity of photosystem I (PSI) using the silver island films (SIFs), gold nanoparticles (AuNP) and ordered monometallic nanostructures, called Fischer patterns. An increase in fluorescence up to 37-fold accompanied by a shift in peak position of fluorescence have been reported [
14,
15,
16]. Recently, an enhancement in fluorescence up to 200-fold for PSI coupled to SIFs at room temperature have also been reported by Czechowski
et al. [
17]. In this article, however, we have explored the effects of coupling PSI with disordered and arbitrary-shaped bimetallic nanostructures. The nanostructure samples are fabricated on commercial glass coverslips by thermal annealing of thin metallic films. The choice of this fabrication technique is made because it is a relatively simple and low cost way of fabrication. In addition, this technique is able to produce large-scale substrates, and optical properties of the nanostructures can be tuned easily [
18,
19]. The fluorescence of PSI deposited on bimetallic nanostructures is investigated using a home-built confocal microscopy setup operating at a cryogenic temperature (1.6 K).
PSI isolated from
Thermosynechococcus elongatus (
Th. elongatus) is used for the investigation, because it is one of the most sophisticated and efficient multi-subunit trans-membrane complexes, responsible for harvesting solar energy to support light-induced charge separations. PSI absorbs over a broad spectral range of the solar radiation reaching the Earth's surface. Irrespective of the wavelength, approximately each absorbed photon by PSI is used to induce electron translocation, which turns over to a quantum yield for charge separation of almost unity [
20]. Such remarkable quantum yield makes PSI a promising candidate for photoelectronic and photochemical devices [
21,
22,
23,
24]. The X-ray structure analysis of PSI from
Th. elongatus resolved at 2.5 Å [
25] revealed that trimeric PSI have a clover leaf structure of 22 nm in diameter [
26]. The monomeric form of PSI from
Th. elongatus is comprised of ∼ 96 chlorophyll (Chl) molecules responsible for light harvesting, excitation energy transfer and charge separation [
25,
27]. The electron transport chain positioned in the center of monomeric PSI consists of a dimer of Chl molecules named P700,
,
and iron-sulfur clusters (
,
and
) [
26,
27]. P700, the reaction center (RC), is the primary electron donor [
26]. After absorption of light by the antenna system, the excitation energy is transferred to P700, where a light-induced charge separation takes place [
27,
28]. The light harvesting antennas in PSI contain the coupled Chls as dimers and trimers, which absorb at a wavelength higher than 700 nm. Some of them are located near the electron transfer chain. Because of their lower site energies than P700, these pigments are also called red Chls or long wavelength Chls (LWCs) [
26,
28,
29,
30]. Although the actual functions of these LWCs are still under discussion, the key roles may lie in increasing the spectral width of light absorption and funneling the excitation energy to the center of the complex [
26]. At room temperature, the energy absorbed by LWCs is transferred to P700 through the uphill energy transfer mechanism. At low temperatures, the transfer of energy to P700 is blocked, and the absorbed energy is trapped by LWCs, which is then partially released in the form of fluorescence [
28].
Single-molecule spectroscopy (SMS) is used to study the fluorescence of the LWCs in the presence of plasmonic nanostructures. The important aspect of SMS is to unravel information hidden in ensemble spectra by avoiding ensemble averaging effects [
31]. At a cryogenic temperature, conformational fluctuations in proteins and their environment are reduced. Therefore, the photo and frequency stability of protein-bound chromophores at lower temperatures increase, and spectroscopic studies with high spectral sensitivity become possible [
32]. Therefore, probing single PSI complexes coupled to plasmonic nanostructures, using SMS below the liquid helium (He) temperature, provides a chance for comprehensive investigation. Based on this detailed investigation, the impacts of the plasmonic nanostructures on the optical properties of PSI are discussed.
2. Experimental Section
Bimetallic nanostructures were fabricated on thin commercial glass coverslips of 20 mm × 20 mm by using the thermal annealing technique. Prior to metal evaporation, the glass substrates were washed with a mixture of detergent (Decon 90) and double-distilled water with a volume ratio of 1:9 in a water bath at 50 °C for 20 min. In order to ensure that all of the detergent residues had been removed, the washing process was repeated twice with double-distilled water in the ultrasonic cleaner, and afterwards, the substrates were dried by using a
stream. The thin layer of metal (2 nm) was deposited by using a vacuum evaporator under a high vacuum of pressure 1.0 × 10
−6 Torr. For fabrication of the Au/Au sample (
i.e., Au-T-Au-T-Au), three successive evaporations of thin gold films were conducted, where each two evaporations were separated by thermal annealing (T). Evaporating the same quantity of gold in three successive evaporations separated by thermal annealing provides denser nanoparticles with a smaller inter-particle distance. Similarly, for fabrication of Ag/Au sample (
i.e., Ag-T-Au), at first 2-nm thin film of silver was deposited followed by thermal annealing and deposition of thin film of gold.
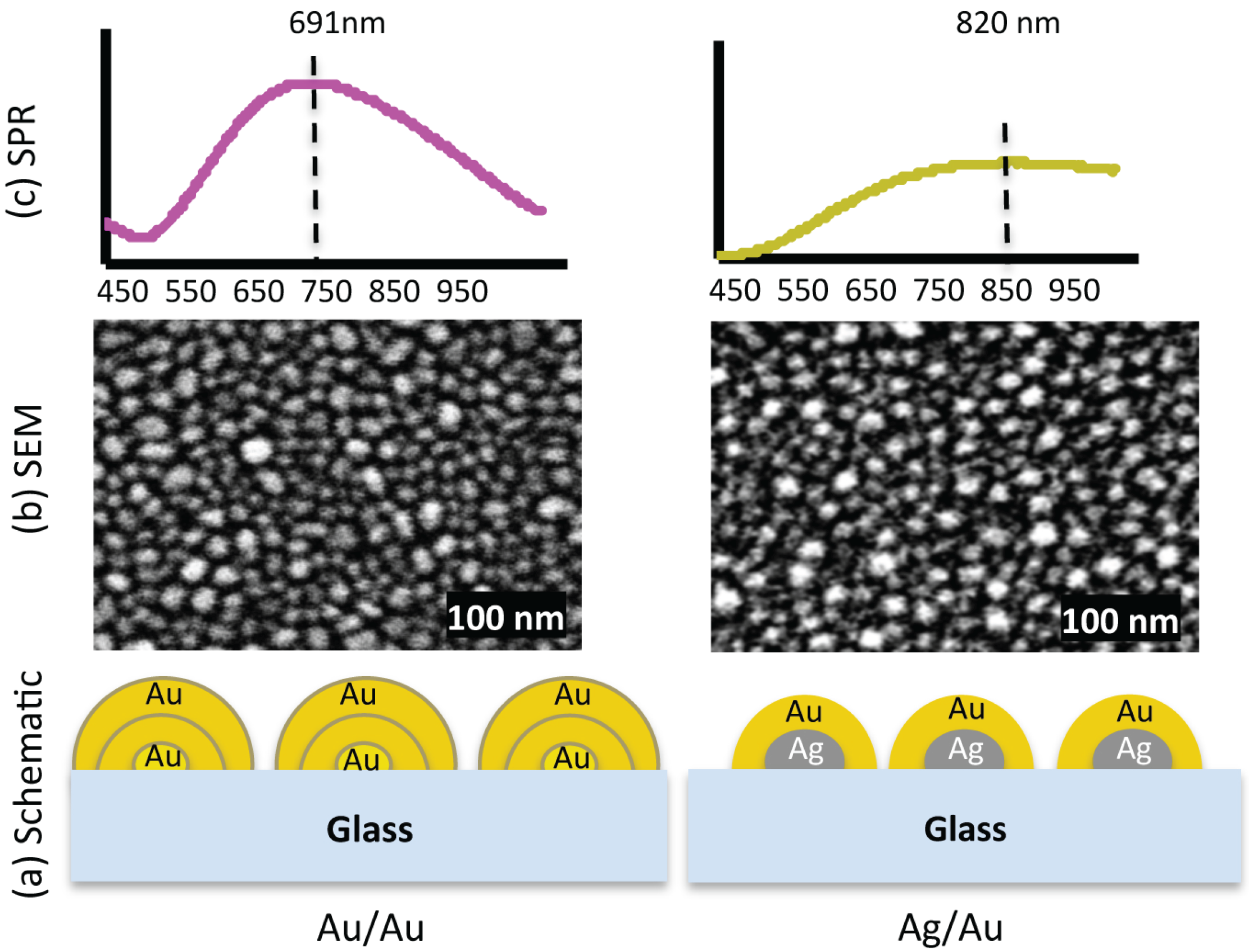
Figure 1a shows a schematic diagram of both of the samples. Every thermal annealing operation was performed for 20 s by using hot plate at 250 °C.
Figure 1.
(a) Exaggerated schematic diagram of two nanostructured substrates, Au/Au and Ag/Au. (b) The SEM images show that the nanostructures are different in shape, size and inter-structure spacing. In the Au/Au sample, the smallest size is ≈ 6 nm and the largest ≈ 47 nm, and for Ag/Au sample, the smallest size is ≈ 6 nm and the largest ≈ 36 nm. (c) SPR spectra of the respective samples. The extinction peaks for samples Au/Au and Ag/Au are at 691 nm and 820 nm, respectively.
Figure 1.
(a) Exaggerated schematic diagram of two nanostructured substrates, Au/Au and Ag/Au. (b) The SEM images show that the nanostructures are different in shape, size and inter-structure spacing. In the Au/Au sample, the smallest size is ≈ 6 nm and the largest ≈ 47 nm, and for Ag/Au sample, the smallest size is ≈ 6 nm and the largest ≈ 36 nm. (c) SPR spectra of the respective samples. The extinction peaks for samples Au/Au and Ag/Au are at 691 nm and 820 nm, respectively.
Trimeric PSI from
Th. elongatus used in this study has been isolated as described in [
33]. The purified PSI trimers were diluted in buffer solution to achieve a low concentration (∼ 3 pM), which is necessary for SMS. The detailed procedure for the preparation of the buffer is explained in [
14]. To form a hybrid, less than 1
μL of this highly diluted solution was sandwiched between metallic nanostructures and bare glass coverslips and transferred to a pre-cooled (4.2 K) cryostat. SMS is performed by using a home-built confocal microscope setup operating at cryogenic temperature ∼1.6 K [
34]. A
cw diode laser at a wavelength of 665 nm was used as an excitation source for fluorescence measurements. In total, 100 spectra were collected for one dataset from one isolated coupled complex by using the excitation power of 100
μW.
3. Results
Irregular bimetallic nanostructures fabricated by thermal annealing of thin metallic films, having different size, shape and inter-structure spacing were used to investigate the influence of plasmonic interaction on optical properties of PSI [
18]. The schematic diagram shown in
Figure 1a is a simplified representation of the composition of two different nanostructures used in our experiments. The fabrication conditions,
i.e. pressure, annealing temperature, annealing timing and film thickness
etc., were kept same for all samples. The nanostructures are disorganised with arbitrary shape. Some of the them are round shape while others are more elongated.
Figure 1b shows the scanning electron microscope (SEM) images of the respective nanostructures. The size of the nanostructures is not same but varying between 6–47 nm in diameter for Au/Au sample and between 6–36 nm for Ag/Au sample. The average diameter of the nanostructures for Au/Au is ≈ 19 nm with standard deviation of 6.1 nm (for particle size distribution see supplementary information), while for Ag/Au sample the average diameter is ≈ 18 nm with standard deviation of 6.4 nm.
Figure 1c shows the surface plasmon resonance (SPR) spectra of these samples. The extinction peaks of Au/Au and Ag/Au samples are at 691 nm and 820 nm, respectively. The extinction spectrum of Au/Au sample is intense and well shaped with defined peak, whereas the extinction spectrum of Ag/Au is broad with low intensity. The differences in extinction spectra of two samples are because of their different metal compositions, size, shape and inter-structure spacing [
18].
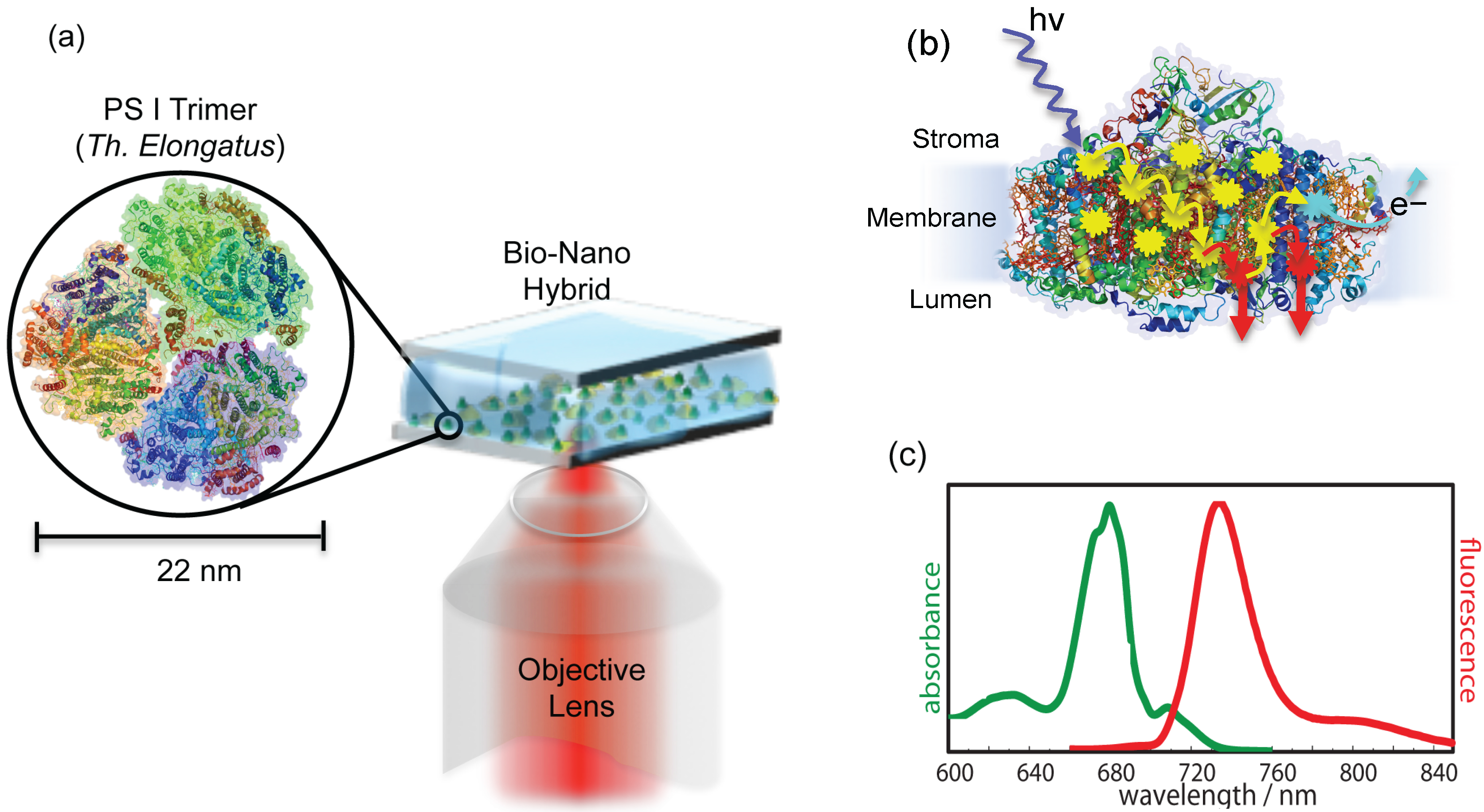
Figure 2.
(
a) Schematic illustration of experimental arrangement. A buffer solution containing isolated and purified photosystem I (PSI) trimers from
is sandwiched between metallic nanostructures and a bare glass coverslip. The green dots represent PSI trimers, while random yellow dots are metallic nanostructures. The inset shows the trimeric PSI from
(protein data bank (PDB) entry: 1JB0). The same objective is used for the excitation and collection of back reflected light. (
b) The side view of PSI in monomeric form with exaggerated details about the excitation energy transfer (EET) pathways. At ambient temperatures, the captured excitation energy is transferred along the transfer chain (shown by yellow arrows) to the reaction center (RC) (represented by cyan explosion symbol), where light-induced charge separation takes place. However, at low temperatures, the energy transfer to the RC is blocked, so instead of charge separation, the captured energy is partially released in the form of fluorescence from red Chls (represented by red arrows). Around 96 Chls per monomer participate in the energy transfer process. (
c) Absorption and emission spectra of ensemble PSI from
at a low temperature (77 K) in the oxidized state, adopted from [
28].
Figure 2.
(
a) Schematic illustration of experimental arrangement. A buffer solution containing isolated and purified photosystem I (PSI) trimers from
is sandwiched between metallic nanostructures and a bare glass coverslip. The green dots represent PSI trimers, while random yellow dots are metallic nanostructures. The inset shows the trimeric PSI from
(protein data bank (PDB) entry: 1JB0). The same objective is used for the excitation and collection of back reflected light. (
b) The side view of PSI in monomeric form with exaggerated details about the excitation energy transfer (EET) pathways. At ambient temperatures, the captured excitation energy is transferred along the transfer chain (shown by yellow arrows) to the reaction center (RC) (represented by cyan explosion symbol), where light-induced charge separation takes place. However, at low temperatures, the energy transfer to the RC is blocked, so instead of charge separation, the captured energy is partially released in the form of fluorescence from red Chls (represented by red arrows). Around 96 Chls per monomer participate in the energy transfer process. (
c) Absorption and emission spectra of ensemble PSI from
at a low temperature (77 K) in the oxidized state, adopted from [
28].
![Photonics 02 00838 g002]()
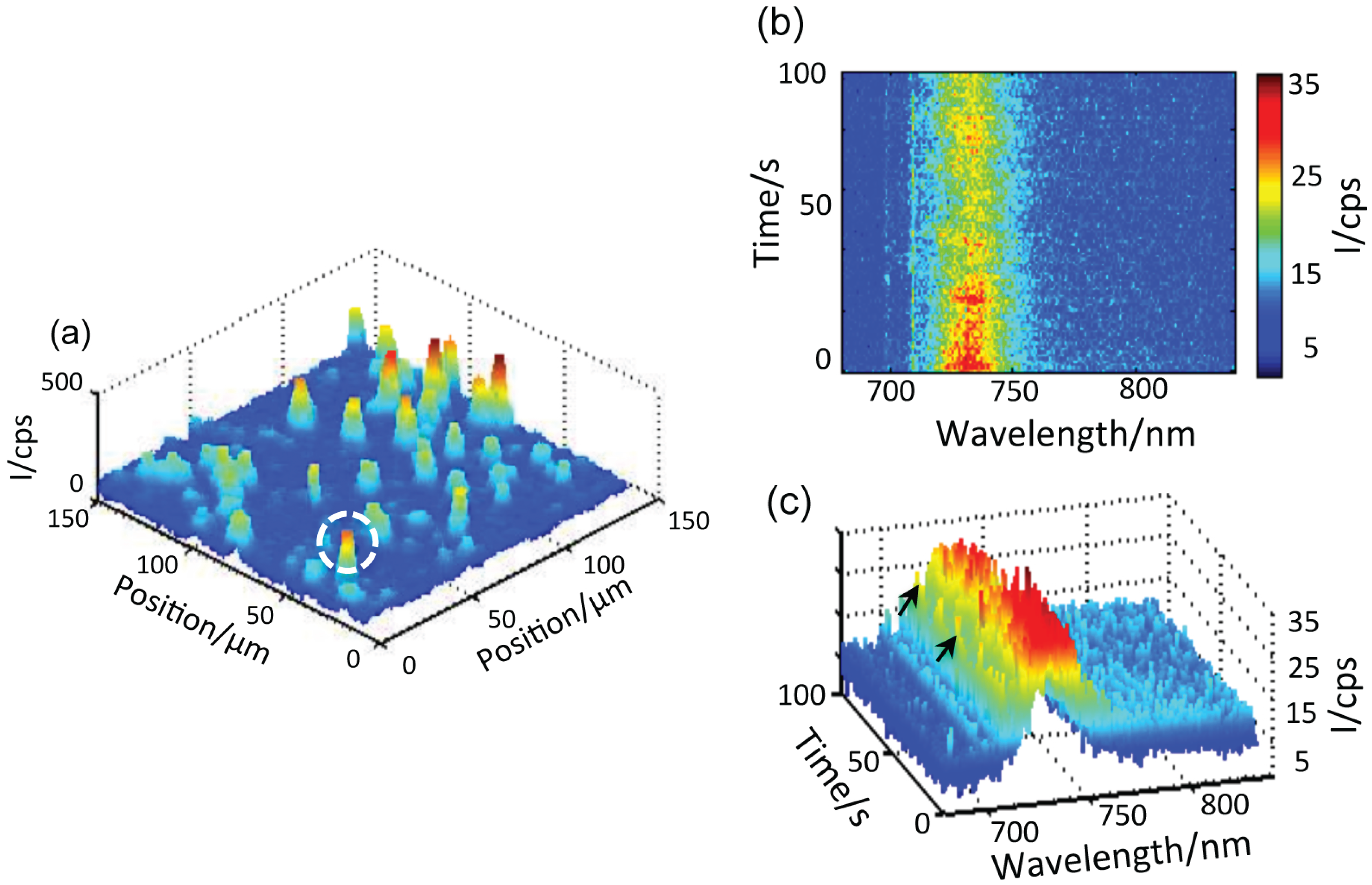
Figure 3.
(a) Confocal fluorescence 3D scan of PSI-Au/Au. The bright spots, which are visible in almost the entire image range, are from individual PSI-Au/Au pairs. The white circle highlights the exemplary individual pair from which the sequence of spectra are collected. (b) The time-dependent dataset consists of 100 fluorescence spectra taken from the highlighted single PSI-Au/Au pair with an integration time of 1 s per spectra. (c) The 3D view of the sequence of 100 spectra.
Figure 3.
(a) Confocal fluorescence 3D scan of PSI-Au/Au. The bright spots, which are visible in almost the entire image range, are from individual PSI-Au/Au pairs. The white circle highlights the exemplary individual pair from which the sequence of spectra are collected. (b) The time-dependent dataset consists of 100 fluorescence spectra taken from the highlighted single PSI-Au/Au pair with an integration time of 1 s per spectra. (c) The 3D view of the sequence of 100 spectra.
These bimetallic nanostructures were coupled to PSI to form the bio-nano hybrids. To achieve the coupling of PSI with nanostructures, a solution of PSI trimers from
was deposited on top of metallic nanostructures, which were then covered with bare glass coverslips, as shown in
Figure 2a. These samples were named PSI-Au/Au and PSI-Ag/Au. An additional sample was prepared as a reference by sandwiching single PSI complexes between two bare glass coverslips and was named PSI-glass. To ensure the observation of individual coupled pairs in the entire width of the excitation beam's focus, a very low concentration of PSI trimers (∼ 3 pM) in the solution was used. The samples were prepared outside of the cryostat and subsequently were inserted quickly onto the scanning desk in the cooled cryostat (4.2 K) by using our fast sample transfer mechanism introduced in [
34]. The samples were scanned in the
xy direction over a specific range.
Figure 3a shows one of the confocal fluorescence 3D scan images taken from the PSI-Au/Au sample. The bright spots that are visible in almost the entire image range are from individual coupled PSI complexes. The deviation of the spots from a circular shape can be attributed to the low image quality of the microscope objective used at low temperature. From each of these coupled PSI complexes, a spectra series consisting of 100 spectra was recorded continuously with an acquisition time of 1 s per spectrum.
Figure 3b shows one example of the time-dependent dataset taken from the highlighted exemplary individual coupled pair of the PSI-Au/Au sample. In
Figure 3c, the 3D view of time-dependent dataset is shown. Notably, intense zero phonon lines (ZPLs) marked with black arrows around 710 nm and a broad emission in the region around 729 nm dominate the whole dataset. The peak position of each individual spectrum can be seen as jumping in a certain wavelength range. Furthermore, photoblinking accompanied with a decay in intensity is obvious from the dataset over time. These spectral features,
i.e., the appearance of zero phonon lines and jumps in the peak position of each spectrum, clearly indicate the fluorescence of a single molecule.
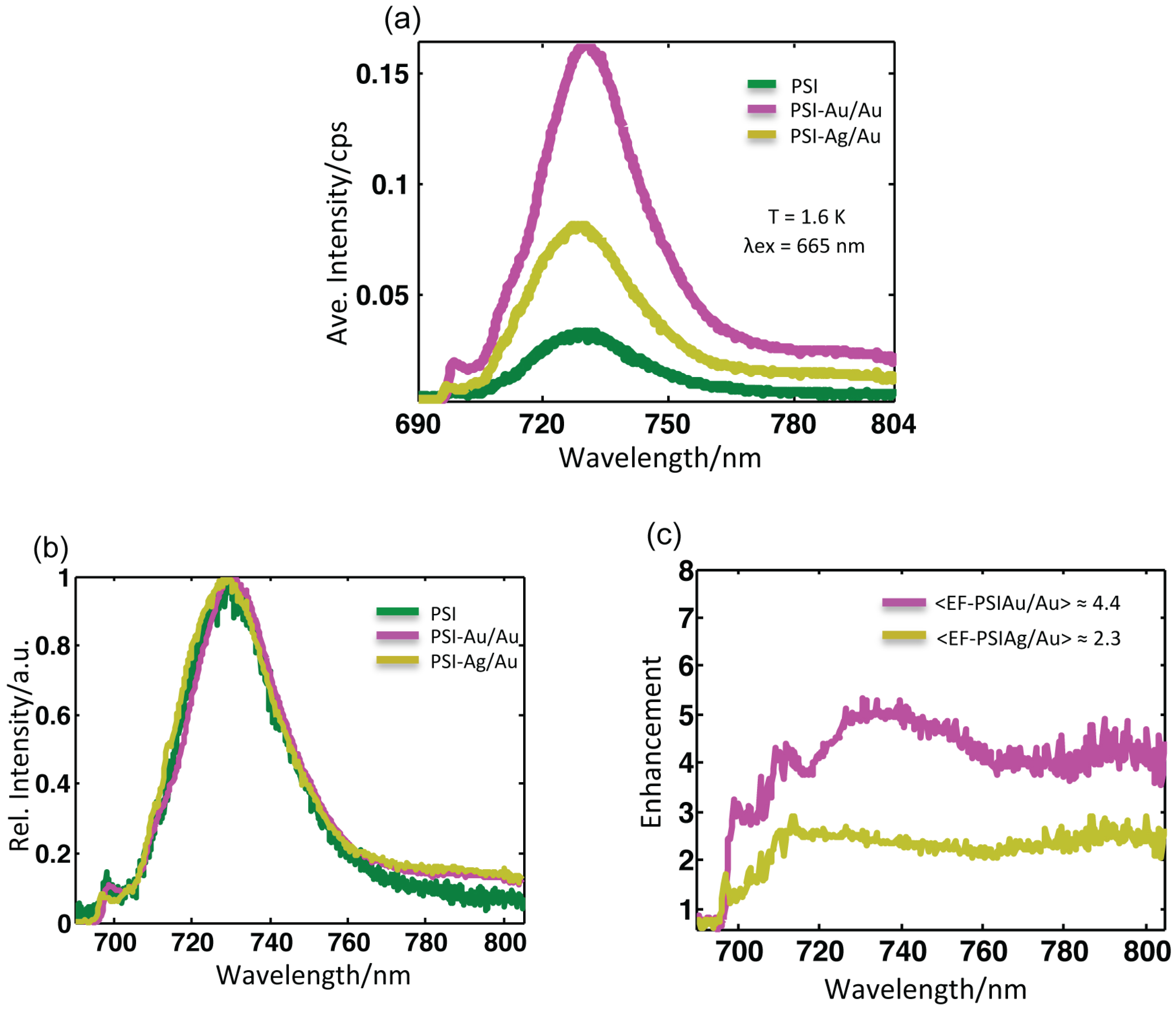
Figure 4.
(a) Average fluorescence spectra of all samples after baseline correction at T = 1.6 K upon excitation at 665 nm. The accumulation time for PSI-glass was 2 s, while for PSI-Au/Au and PSI-Ag/Au, it was 1 s. (b) The normalized average spectra were scaled to the same maximum value after baseline correction. No pronounced broadening in the linewidth of any sample was found. (c) Wavelength-dependent enhancement spectra obtained after dividing the average spectra of PSI-Au/Au and PSI-Ag/Au by the average spectrum of PSI-glass. More enhancement was observed around main peak region (730 nm) and in the far red region (> 780 nm).
Figure 4.
(a) Average fluorescence spectra of all samples after baseline correction at T = 1.6 K upon excitation at 665 nm. The accumulation time for PSI-glass was 2 s, while for PSI-Au/Au and PSI-Ag/Au, it was 1 s. (b) The normalized average spectra were scaled to the same maximum value after baseline correction. No pronounced broadening in the linewidth of any sample was found. (c) Wavelength-dependent enhancement spectra obtained after dividing the average spectra of PSI-Au/Au and PSI-Ag/Au by the average spectrum of PSI-glass. More enhancement was observed around main peak region (730 nm) and in the far red region (> 780 nm).
Figure 4a shows the corresponding mean intensity spectra of all of the datasets from samples PSI-glass (green), PSI-Au/Au (magenta) and PSI-Ag/Au (yellow). The emission profiles of these samples with normalized intensity are shown in
Figure 4b. The profile of PSI-Au/Au is similar to PSI-Ag/Au; however, the intensity enhancement is different. An enormous increase in the mean intensity of PSI-Au/Au and PSI-Ag/Au samples is evident. The full width at half maximum (FWHM) of the average spectra for PSI-glass, PSI-Au/Au and PSI-Ag/Au is 27.6, 27.3 and 28.3 nm, respectively.
Table 1 shows the comparison of different values for the different samples. The change in FWHM due to metallic nanostructures in the neighborhood is quite small. A slight increase in FWHM for the PSI-Ag/Au sample as compared to PSI-glass, while a minor decrease for PSI-Au/Au are spotted. The spectral position of emission maxima for PSI-glass was found at 730.4 nm, whereas for PSI-Au/Au with a slight red shift at 731.3 nm and for PSI-Ag/Au with a slight blue shift at 729.7 nm. Compared to the profile of PSI-glass, an additional broad shoulder in the emission profile of PSI-Au/Au and PSI-Ag/Au in the far red region between 780–850 nm is detected. Analogous to the main peaks, the additional shoulder of the PSI-Au/Au sample was also more intense compared to the PSI-Ag/Au sample.
Table 1.
Comparison of enhancement factors, linewidths and standard deviations of enhancement factors observed for uncoupled and coupled PSI complexes.
Table 1.
Comparison of enhancement factors, linewidths and standard deviations of enhancement factors observed for uncoupled and coupled PSI complexes.
| Sample | PSI-glass | PSI-Au/Au | PSI-Ag/Au |
|---|
| Emission Maximum (nm) | 730.4 | 731.3 | 729.7 |
| FWHM (nm) | 27.6 | 27.3 | 28.3 |
| Average Enhancement | - | 4.4 | 2.3 |
| Max. Enhancement | - | 22.9 | 5.1 |
| Standard Deviation | 0.3 | 3.9 | 0.8 |
The wavelength-dependent enhancement in intensities of PSI-Au/Au and PSI-Ag/Au samples are plotted at 690–805 nm after dividing by the average spectra of PSI-glass. The resulting wavelength-dependent enhancement spectra are shown in
Figure 4c. For the PSI-Au/Au sample, a continuous fluorescence enhancement with three intensified domains,
i.e., one broad one in the region at 720–760 nm and two relatively narrow domains,
i.e. at 706–716 nm and at 780–805 nm, was recorded. A similar trend of continuous enhancement was seen for the PSI-Ag/Au sample, but intensity enhancement was comparatively decreased as compared to the PSI-Au/Au sample. Furthermore, there was only one prominent domain at 780–805 nm with a comparatively high intensity. It is worth noting that, because of the dependence of the selected procedure on the elimination of background contributions, the risk of error in a spectral region with low intensity becomes higher compared to the high intensity spectral region.
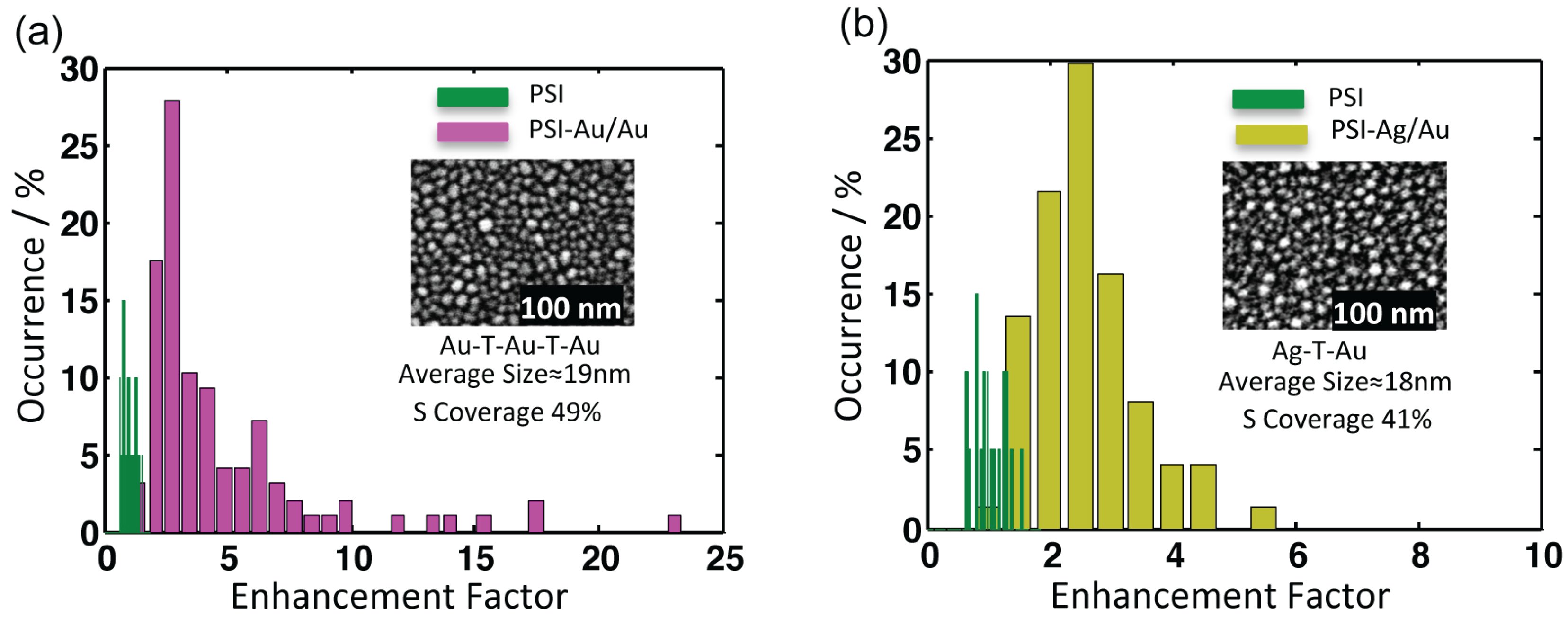
All nanostructures lead to an emission enhancement of coupled PSI complexes. For calculating enhancement factors, the intensity counts from individual datasets of PSI-Au/Au and PSI-Ag/Au samples were integrated after baseline correction and compared with average intensity counts of PSI-glass. The distribution of the relative intensities from individual complexes of PSI-Au/Au and PSI-Ag/Au with respect to PSI-glass are displayed in
Figure 5. The relative intensity of individual datasets of PSI-Au/Au and PSI-Ag/Au samples with respect to the mean intensity of PSI-glass is referred to as the enhancement factor. The intensity histogram of PSI-glass (green) shows almost a Gaussian shape, while the histograms of the enhancement factors for PSI-Au/Au and PSI-Ag/Au initially increase steeply to a maximum value and decay approximately exponentially afterwards. The insets are the SEM micrographs of metallic nanostructures with information about their respective fabrication methods, surface coverage and the size of the nanostructures. For PSI-Au/Au, the average increase in fluorescence intensity is 4.4 ± 3.9-fold, while for PSI-Ag/Au, it increases by 2.3 ± 0.8-fold. However, for individual datasets, the maximum enhancement factors for PSI-Au/Au and PSI-Ag/Au samples are 22.9 and 5.1, respectively. The average enhancement factors for both PSI-Au/Au and PSI-Ag/Au samples are higher than those previously reported for hybrid of PSI with ordered gold Fischer patterns (PSI-AuΔ) [
14].
Figure 5.
Comparison of the integrated fluorescence intensities of (a) PSI-Au/Au (magenta) and (b) PSI-Ag/Au (yellow) with PSI-glass in green. The insets are the SEM images of the respective nanostructures. The counts of individual datasets were integrated after baseline correction. The average enhancement factors for PSI-Au/Au and PSI-Ag/Au are 4.4 ± 3.9 and 2.3 ± 0.8, respectively. The x-axis defines the enhancement factor, whereas the y-axis shows the relative percentage of occurrence of the datasets with respective enhancement factors.
Figure 5.
Comparison of the integrated fluorescence intensities of (a) PSI-Au/Au (magenta) and (b) PSI-Ag/Au (yellow) with PSI-glass in green. The insets are the SEM images of the respective nanostructures. The counts of individual datasets were integrated after baseline correction. The average enhancement factors for PSI-Au/Au and PSI-Ag/Au are 4.4 ± 3.9 and 2.3 ± 0.8, respectively. The x-axis defines the enhancement factor, whereas the y-axis shows the relative percentage of occurrence of the datasets with respective enhancement factors.
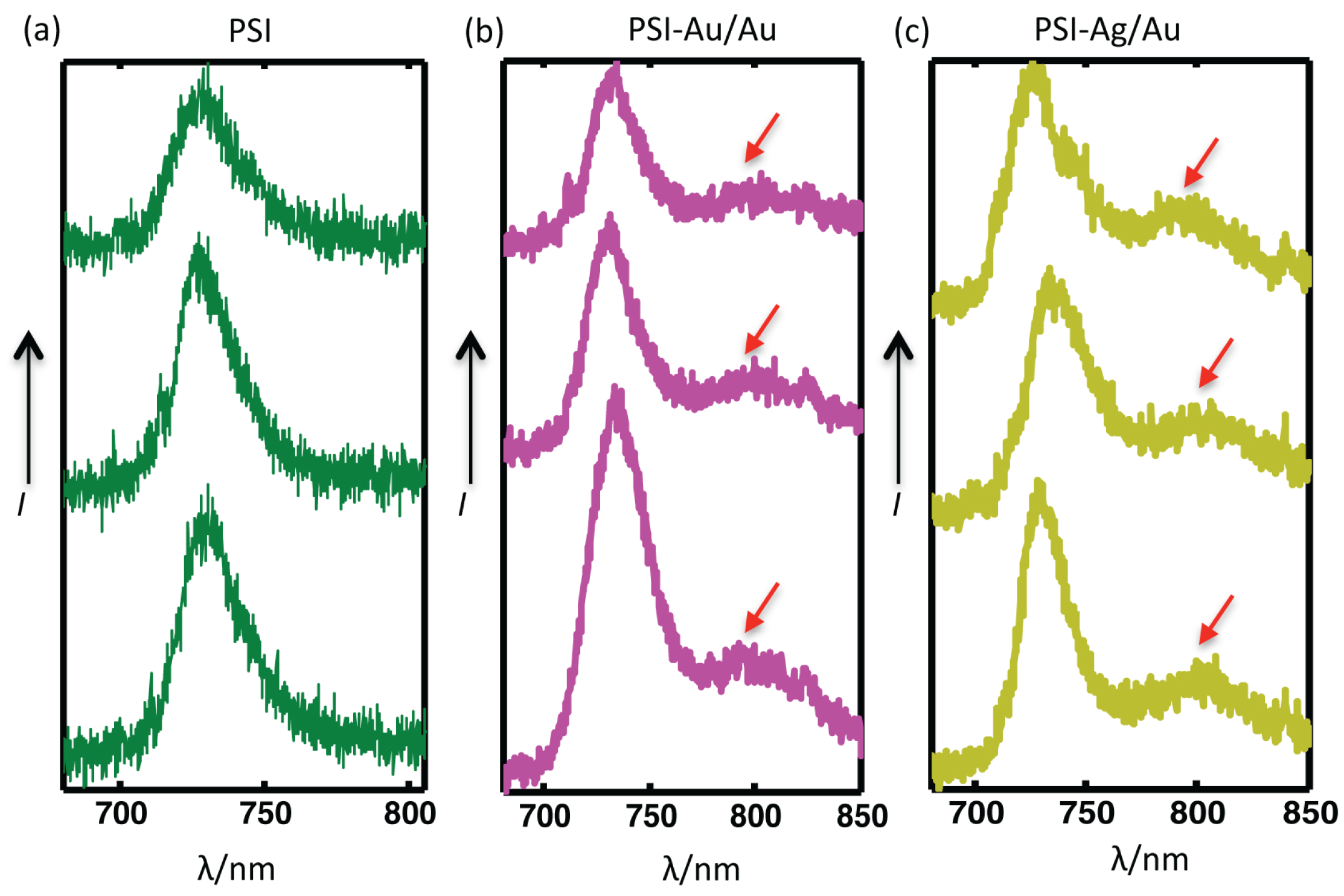
Figure 6.
Comparison of three individual datasets from three single complexes of each (a) PSI-glass (b) PSI-Au/Au and (c) PSI-Ag/Au. The red arrows point to the broad enhanced shoulders appearing in the far red region (from 780–850 nm) for almost all coupled complexes of PSI-Au/Au and PSI-Ag/Au samples. The accumulation time for each PSI was 2 s, while for PSI-Au/Au and PSI-Ag/Au, it was 1 s.
Figure 6.
Comparison of three individual datasets from three single complexes of each (a) PSI-glass (b) PSI-Au/Au and (c) PSI-Ag/Au. The red arrows point to the broad enhanced shoulders appearing in the far red region (from 780–850 nm) for almost all coupled complexes of PSI-Au/Au and PSI-Ag/Au samples. The accumulation time for each PSI was 2 s, while for PSI-Au/Au and PSI-Ag/Au, it was 1 s.
The comparison of the spectra from three individual complexes of each, PSI-glass, PSI-Au/Au and PSI-Ag/Au, are shown in
Figure 6. Clear, distinctive and broad enhanced shoulders, which appeared for both of the PSI-Au/Au and PSI-Ag/Au samples in the wavelength range 780–850 nm, are elaborated. The red arrows point out these broad shoulders, which were consistent in all datasets of PSI-Au/Au and PSI-Ag/Au with varying intensity; while such shoulders have not been detected for PSI-glass.
4. Discussion
The fluorescence enhancement of bio-molecules due to metal nanostructures or nanoparticles in close vicinity has been reported before [
14,
15,
16,
35,
36]. The aforementioned results have shown an increased fluorescence of PSI trimers induced by the plasmonic nanostructures Au/Au and Ag/Au, fabricated by the thermal annealing process. Recent studies have revealed that the spectral overlap between absorption spectra of the fluorophore and extinction spectra from metallic nanoparticles [
37,
38,
39], their mutual distance [
40], relative orientation [
41] and peak position of SPR of metallic nanoparticles [
37,
42] play critical roles in changing the fluorescence of the fluorophores. Therefore, the nanostructures having different SPR peak positions and covering a broad range of wavelengths were fabricated. The SPR peak positions of the nanostructures are tuned by adjusting the fabrication conditions, e.g., metallic film thickness, annealing temperature, time for annealing,
etc. [
18]. The background signals of these nanostructures are negligibly small as compared to the background signals of spherical AuNPs or SIFs [
14,
16]. Therefore, the data evaluations for PSI-Au/Au and PSI-Ag/Au samples are easier compared to PSI-AuNPs and PSI-SIFs [
15]. Any analysis to separate the signals emanating from PSI or from nanostructures is not necessary.
Both of the plasmonic nanostructures used for the investigation yield an enhancement in the fluorescence of PSI. Although the enhancements of fluorescence intensity of PSI-Au/Au and PSI-Ag/Au are lower compared to PSI-AuNPs and PSI-SIFs [
15], the low auto-luminescence of these structures makes them the preferred tool for spectroscopy [
16]. On average, the integral fluorescence intensity of PSI-Au/Au increases by a factor of 4.4 ± 3.9 compared to PSI-glass. For the PSI-Ag/Au sample, the corresponding value is 2.3 ± 0.8, as can be seen in
Figure 4. However, it is worthwhile to note that for the individual complex of PSI-Au/Au, we even observed an increase in intensity of fluorescence up to 22.9-fold and for the PSI-Ag/Au sample up to 5.1-fold (
Figure 5). The average enhancement factor due to Au/Au nanostructures is almost two-times bigger than the ordered gold triangles (AuΔ); however, due to the Ag/Au nanostructures, the enhancement factor is comparable to the results reported for ordered AuΔ [
14,
16]. The difference in enhancement factors of PSI-Au/Au and PSI-Ag/Au is mainly attributed to the difference of the spectral overlap between the absorption of PSI and the extinction spectra of Au/Au and Ag/Au nanostructures [
39]. The effect of plasmonic nanostructures on changing the fluorescence (
i.e., quenching or enhancing) of fluorophore strongly depends on the spectral overlap of plasmon resonance, the absorption spectrum of the fluorophore and excitation wavelength [
37,
39,
43]. The fluorescence responses of proteins have shown that the enhanced fluorescence was observed for the excitation in the spectral zone of metallic nanostructures [
39]. Furthermore, for dyes, the brightest fluorescence is reported, where the emission peak of dyes is slightly red shifted to the localized surface plasmon resonance (LSPR) peak of nanoparticles [
37,
42]. For the PSI-Au/Au sample, the excitation of PSI is within the spectral zone of the Au/Au sample's plasmon resonance, and also the extinction spectrum is more intensive and narrow, with the SPR peak position blue shifted to the emission peak of PSI. Therefore, the maximum enhancement of fluorescence complies with the reported results of Beyer
et al. [
39]. On the other hand, the amount of overlap between extinction spectra of Ag/Au and absorption of PSI is less than the overlap between extinction spectra of Au/Au and absorption of PSI, and the peak position of the SPR is red shifted to the emission peak position of PSI trimer from
, causing less enhancement of the fluorescence.
The histograms in
Figure 5 show that the distributions of the intensity values for individual complexes of PSI-Au/Au and PSI-Ag/Au samples are more heterogeneously distributed than PSI-glass. The standard deviations of intensity distributions for PSI-glass, PSI-Au/Au and PSI-Ag/Au samples are presented in
Table 1. The high standard deviation of enhancement factors of PSI-Au/Au and PSI-Ag/Au samples can be because of: (i) heterogeneity in the size of the metallic nanostructures [
44]; (ii) the distance dependence of the interaction between plasmonic nanostructures and fluorophores [
40]; and (iii) different emission/excitation dipole orientations. The size of individual metallic nanostructures used in our experiments is not homogenous, but it varies between 6–47 nm for the Au/Au substrate and between 6–36 nm for the Ag/Au substrate, as can be seen by the SEM images in
Figure 1 (the size distribution of the nanostructures is given in [
19]). Simulations and theoretical studies have shown that with the increase of the size of the particles, the electric field gets more intense and also extends further out from the particle [
45]. Depending on the strength of the field, the fluorescence enhancement varies. Applying the same analogy, broad variation in the size of nanostructures produces a broad distribution of the near-field of nanostructures. As a result, the high standard deviations of the fluorescence enhancement factors are observed for Au/Au and Ag/Au samples (
Figure 5). Distance-dependent enhancement and quenching of the fluorescence of fluorophores was observed by Anger
et al. [
40]. They calculated a distance-dependent enhancement curve for a fluorophore coupled to AuNP and found that the fluorescence is quenched for distances shorter than approximately 2 nm and reaches the maximum value at a 5-nm distance. At a distance higher than 5 nm, the fluorescence of the coupled fluorophore decayed exponentially, leading to vanishing enhancement around 80 nm, suggesting the absence of any coupling between the nanoparticle and the fluorophore. PSI used for the investigation in this study is a protein complex of a diameter of around 22 nm with a clover-shaped structure composed of around 288 Chl molecules in its trimeric form [
26]. After hybridization with metallic nanostructures, the distance dependence of fluorescence plays its part. As some of the emitter Chls are in the distance range for maximum enhancement, while others are in the range for quenching of fluorescence, so the ultimate enhancement factors will be different for different coupled complexes, resulting in the broad distribution of the enhancement factor.
The influence of the plasmonic interactions on the functionality of photosynthetic proteins was studied by Govorov
et al. [
22] and Krassen
et al. [
46]. They established that PSI complexes maintain their charge-transfer and light harvesting capability close to nanoparticles. Based on the literature, the modifications in the structure of PSI complexes due to nanostructures in the proximity seems improbable [
15,
23,
46,
47,
48,
49]. Analogous to the observations of single-chromophore systems and two-chromophore FRET-coupled systems [
35,
44,
50], the PSI coupled to the plasmonic nanostructures Au/Au and Ag/Au show almost a uniform enhancement of fluorescence with a slight deviation in the shape of emission spectra compared to the uncoupled PSI. No pronounced difference around the main peak region in the emission profiles was noticed. The linewidths and fluorescence emission peak positions of the PSI-Au/Au and PSI-Ag/Au samples are nearly identical to PSI-glass, and thus, the average spectra of PSI-Au/Au and PSI-Ag/Au are approximately the same as the average spectrum of PSI-glass (
Figure 4a,b). The resulting values of FWHM and fluorescence peak positions are reported in
Table 1. The observations we made have shown that the enhancement in fluorescence and the deviation in the shape of the emission spectra go hand in hand. For the interaction of PSI with AuNPs and SIFs, where a much higher enhancement in fluorescence was observed, the deviation in the shape of the emission spectra was also very strong [
14,
15,
16]. While for PSI-Au/Au and PSI-Ag/Au samples, we observed less enhancement, that may be the reason for the less deviation in the shape of the emission spectra.
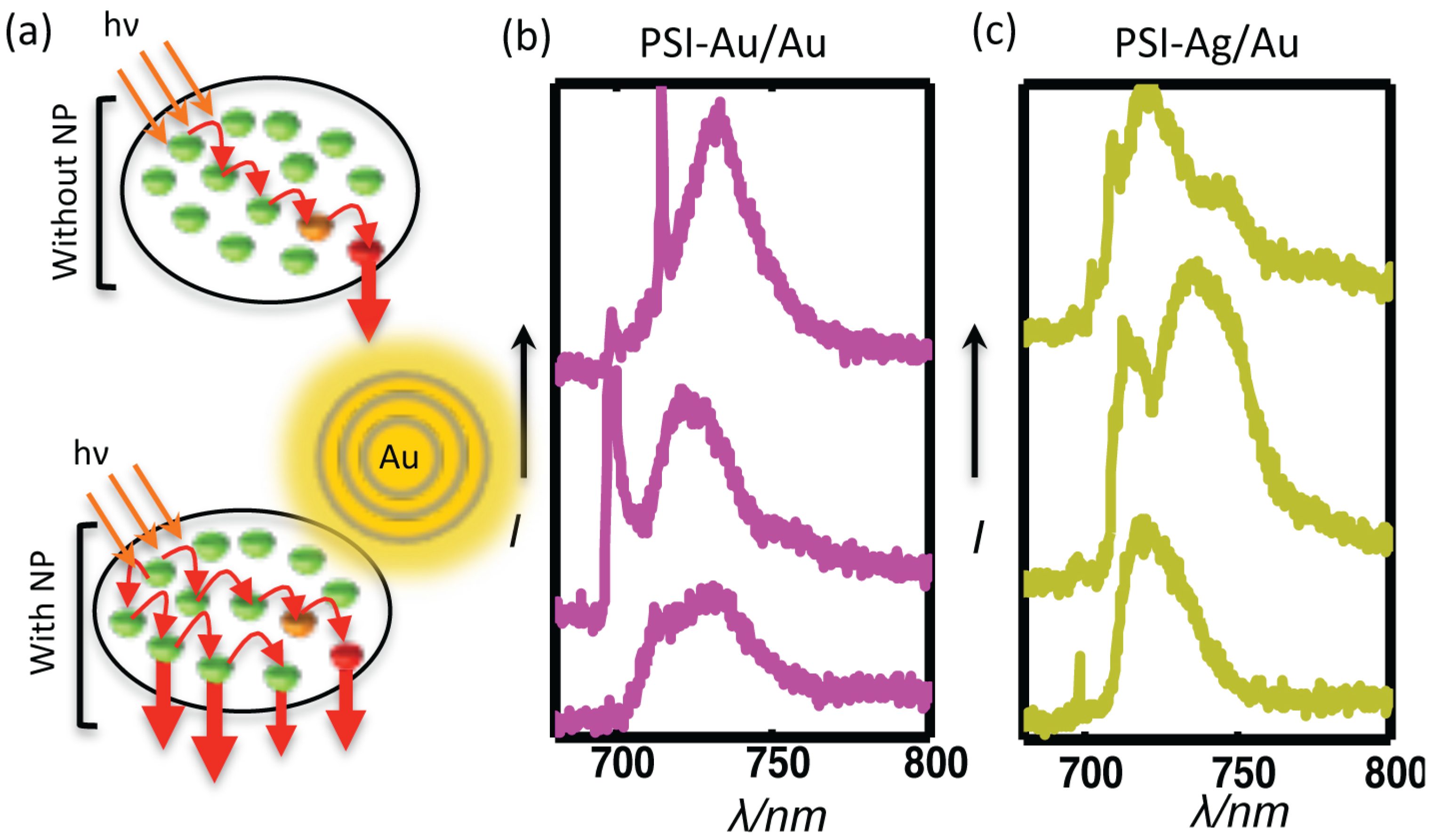
The energy transfer rates among the chromophores of photosynthetic pigment proteins depend on the spectral overlap, spatial separation and orientation of the involved chromophores [
51]. The excitation energy captured by the antenna system is transferred to the RC through chromophores. For the fast and efficient transfer of the excitation energy, the chromophores of the light harvesting system are coupled together [
52]. The specific coupling conditions between the chromophores lead to a characteristic set of transition rates and, thus, to preferred energy transfer pathways [
51,
53]. The effect of plasmonic interaction on the Förster interaction distance of the coupled chromophores was studied by Zhang
et al. [
54]. They published that the Förster radius between chromophores changes due to the interaction with plasmonic nanoparticles depending on the size of the particle and the distance from the particle [
54,
55]. For a single donor-acceptor pair, an increase in Förster radius is proclaimed from 8.3–13 nm [
54]. Using a similar analogy for PSI, it can be speculated that the coupling between PSI-bound chromophores will also change due to the presence of metallic nanostructures leaving effects on the energy transfer efficiencies. As a result, the distribution of the exciton on the chromophores of PSI is affected. The chromophores, which in their native form do not participate in EET, will additionally get involved in the process. Additional EET pathways, as illustrated in
Figure 7a, will be constituted. Moreover, the chromophores that were initially non-fluorescent also become fluorescent; therefore, the fluorescence emission is enhanced [
14,
15]. Variations in the shape of the emission spectra of PSI-Au/Au and PSI-Ag/Au shown in
Figure 7 strengthen this hypothesis.
Along with the main emission band at 729.7 ± 2 nm, we have also noticed enhanced fluorescence shoulders at > 770 nm for both PSI-Au/Au and PSI-Ag/Au samples (
Figure 6). These shoulders in the far red spectral region can be attributed to “vibronic sidebands corresponding to the mirror image of the
(0→1) absorption band of LWC C708” [
28]. In oxidation form P
, the fluorescence intensity on the long-wavelength side of the main emission peak is quenched [
28], because P
has a broad absorption band that shows a considerable overlap with the fluorescence spectrum of LWCs [
56,
57]. As a consequence, the fluorescence of LWCs should reduce; however, contrary to that, we observed an enhancement in this region. This gain in fluorescence intensity can therefore only be explained by considering a dramatic change in the process of fluorescence quenching by P
due to the presence of metallic nanostructures [
14]. Therefore, we assume that metallic nanostructures may have changed the irreversible energy transfer pathways from red Chls to P
, resulting in the lower quenching of fluorescence.
Figure 7.
(a) A simplified illustration of the proposed mechanism for fluorescence enhancement of PSI due to plasmonic nanostructures in the locality. Due to the specific coupling among Chls in the native form of PSI, energy is transferred to the RC by following certain energy transfer pathways. In the presence of plasmonic nanostructures, the coupling among Chls is changed, resulting in the emergence of new energy transfer pathways and the turning on of non-fluorescent Chls, as well. The representative emission spectra of individual coupled complexes for each of the sample types (b) PSI-Au/Au and (c) PSI-Ag/Au show the deviation in their shapes compared to the shape of PSI-glass, indicating the emergence of new energy transfer pathways.
Figure 7.
(a) A simplified illustration of the proposed mechanism for fluorescence enhancement of PSI due to plasmonic nanostructures in the locality. Due to the specific coupling among Chls in the native form of PSI, energy is transferred to the RC by following certain energy transfer pathways. In the presence of plasmonic nanostructures, the coupling among Chls is changed, resulting in the emergence of new energy transfer pathways and the turning on of non-fluorescent Chls, as well. The representative emission spectra of individual coupled complexes for each of the sample types (b) PSI-Au/Au and (c) PSI-Ag/Au show the deviation in their shapes compared to the shape of PSI-glass, indicating the emergence of new energy transfer pathways.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


