Takotsubo as Initial Manifestation of Non-Myopathic Cardiomyopathy Due to the Titin Variant c.1489G > T


{kind=link}
Abstract
:1. Novel Insights
2. Established Facts
3. Introduction
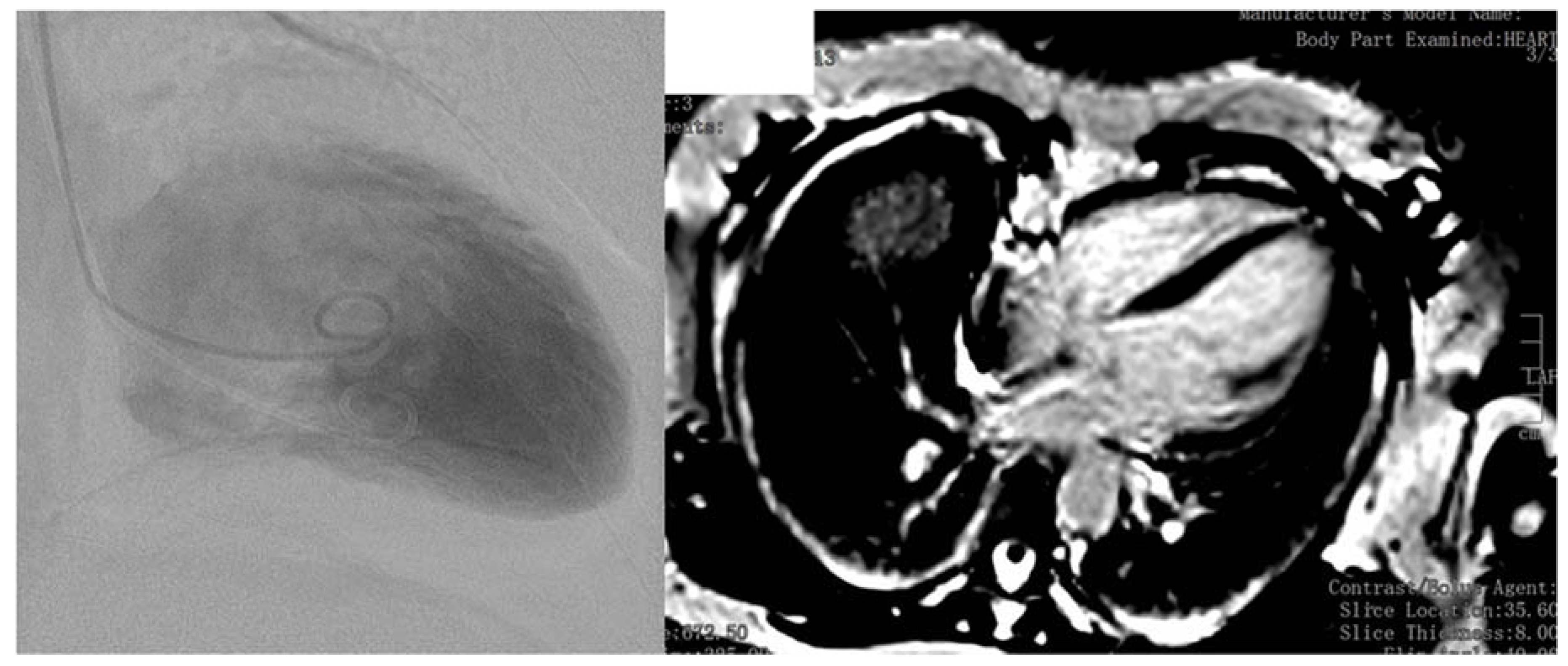
4. Case Report
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Shams, Y.; Tornvall, P. Epidemiology, pathogenesis, and management of takotsubo syndrome. Clin. Auton. Res. 2017, 28, 53–65. [Google Scholar] [CrossRef]
- Tayal, U.; Prasad, S.; Cook, S.A. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knöll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Anastasi, M.; Greif, M.; Reiser, M.F.; Theisen, D. Magnetic resonance imaging of dilated cardiomyopathy. Radiologe 2013, 53, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B. The Rapidly Evolving Role of Titin in Cardiac Physiology and Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.E.; Harris, E.; Barresi, R.; Marsh, J.; Beattie, A.; Bourke, J.P.; Straub, V.; Chinnery, P.F. Cardiac involvement in hereditary myopathy with early respiratory failure: A cohort study. Neurology 2016, 87, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.; Töpf, A.; Vihola, A.; Evilä, A.; Barresi, R.; Hudson, J.; Hackman, P.; Herron, B.; MacArthur, D.; Lochmüller, H.; et al. A ‘second truncation’ in TTN causes early onset recessive muscular dystrophy. Neuromuscul. Disord. 2017, 27, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; Huttner, I.G. Titin-truncating mutations in dilated cardiomyopathy: The long and short of it. Curr. Opin. Cardiol. 2017, 32, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Franaszczyk, M.; Chmielewski, P.; Truszkowska, G.; Stawinski, P.; Michalak, E.; Rydzanicz, M.; Sobieszczanska-Malek, M.; Pollak, A.; Szczygieł, J.; Kosinska, J.; et al. Titin Truncating Variants in Dilated Cardiomyopathy—Prevalence and Genotype-Phenotype Correlations. PLoS ONE 2017, 12, e0169007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, H.; Wu, G.; Luo, X.; Zhang, C.; Zou, Y.; Wang, H.; Hui, R.; Wang, J.; Song, L. Titin-Truncating Variants Increase the Risk of Cardiovascular Death in Patients with Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2017, 33, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) Variants in Chemotherapy-Induced Cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Hastings, R.; de Villiers, C.P.; Hooper, C.; Ormondroyd, L.; Pagnamenta, A.; Lise, S.; Salatino, S.; Knight, S.J.; Taylor, J.C.; Thomson, K.L.; et al. Combination of Whole Genome Sequencing, Linkage, and Functional Studies Implicates a Missense Mutation in Titin as a Cause of Autosomal Dominant Cardiomyopathy with Features of Left Ventricular Noncompaction. Circulation 2016, 9, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12. [Google Scholar] [PubMed]
- Dabby, R.; Sadeh, M.; Hilton-Jones, D.; Plotz, P.; Hackman, P.; Vihola, A.; Udd, B.; Leshinsky-Silver, E. Adult onset limb-girdle muscular dystrophy—A recessive titinopathy masquerading as myositis. J. Neurol. Sci. 2015, 351, 120–123. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, H.; Neuhold, U.; Weidinger, F.; Gatterer, E.; Stöllberger, C.; Huber, K.; Finsterer, J. Takotsubo as Initial Manifestation of Non-Myopathic Cardiomyopathy Due to the Titin Variant c.1489G > T. Medicines 2018, 5, 80. https://doi.org/10.3390/medicines5030080
Keller H, Neuhold U, Weidinger F, Gatterer E, Stöllberger C, Huber K, Finsterer J. Takotsubo as Initial Manifestation of Non-Myopathic Cardiomyopathy Due to the Titin Variant c.1489G > T. Medicines. 2018; 5(3):80. https://doi.org/10.3390/medicines5030080
Chicago/Turabian StyleKeller, Hans, Ulrike Neuhold, Franz Weidinger, Edmund Gatterer, Claudia Stöllberger, Klaus Huber, and Josef Finsterer. 2018. "Takotsubo as Initial Manifestation of Non-Myopathic Cardiomyopathy Due to the Titin Variant c.1489G > T" Medicines 5, no. 3: 80. https://doi.org/10.3390/medicines5030080
APA StyleKeller, H., Neuhold, U., Weidinger, F., Gatterer, E., Stöllberger, C., Huber, K., & Finsterer, J. (2018). Takotsubo as Initial Manifestation of Non-Myopathic Cardiomyopathy Due to the Titin Variant c.1489G > T. Medicines, 5(3), 80. https://doi.org/10.3390/medicines5030080