Glucosamine Enhances TRAIL-Induced Apoptosis in the Prostate Cancer Cell Line DU145

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemical Compounds and Biological Reagents
2.2. Immunoblot Analysis
2.3. Quantitative RT PCR
2.4. Evaluation of DR4 and DR5 TRAIL Receptors by Flow Cytometry
2.5. Cytosolic and Nuclear Protein Extraction
2.6. Detection of Apoptosis by Flow Cytometry Analysis
2.7. Confocal Microscopy
2.8. Caspase 8 Activity Assay
2.9. Statistical Analyses
3. Results
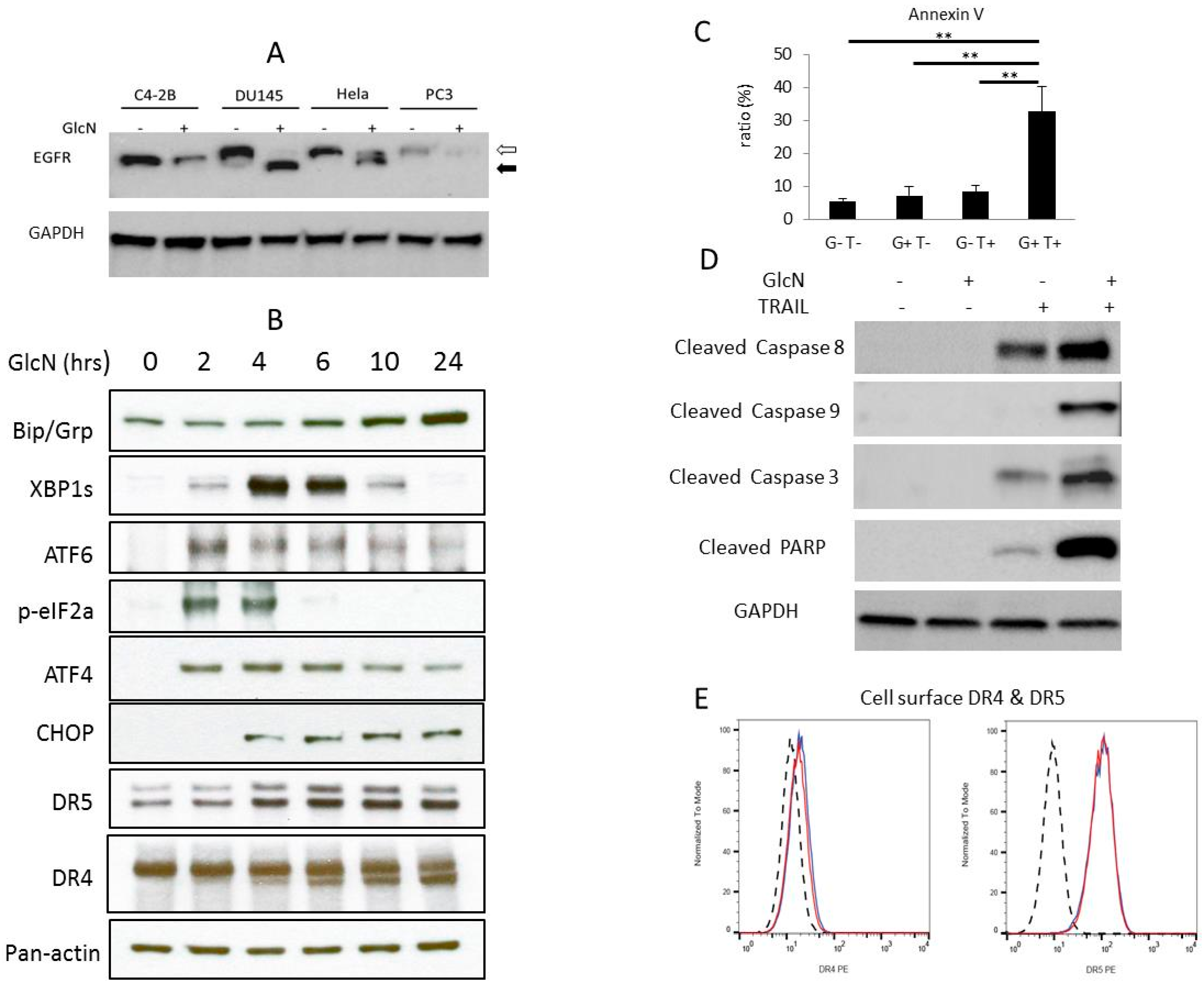
3.1. GlcN Induces ER Stress and Increases Cellular DR5 Expression in DU145 Cells
3.2. GlcN Potentiates TRAIL-Induced Apoptosis in DU145 Cells
3.3. GlcN Does Not Alter the DR4 or DR5 Cell Surface Expression Level
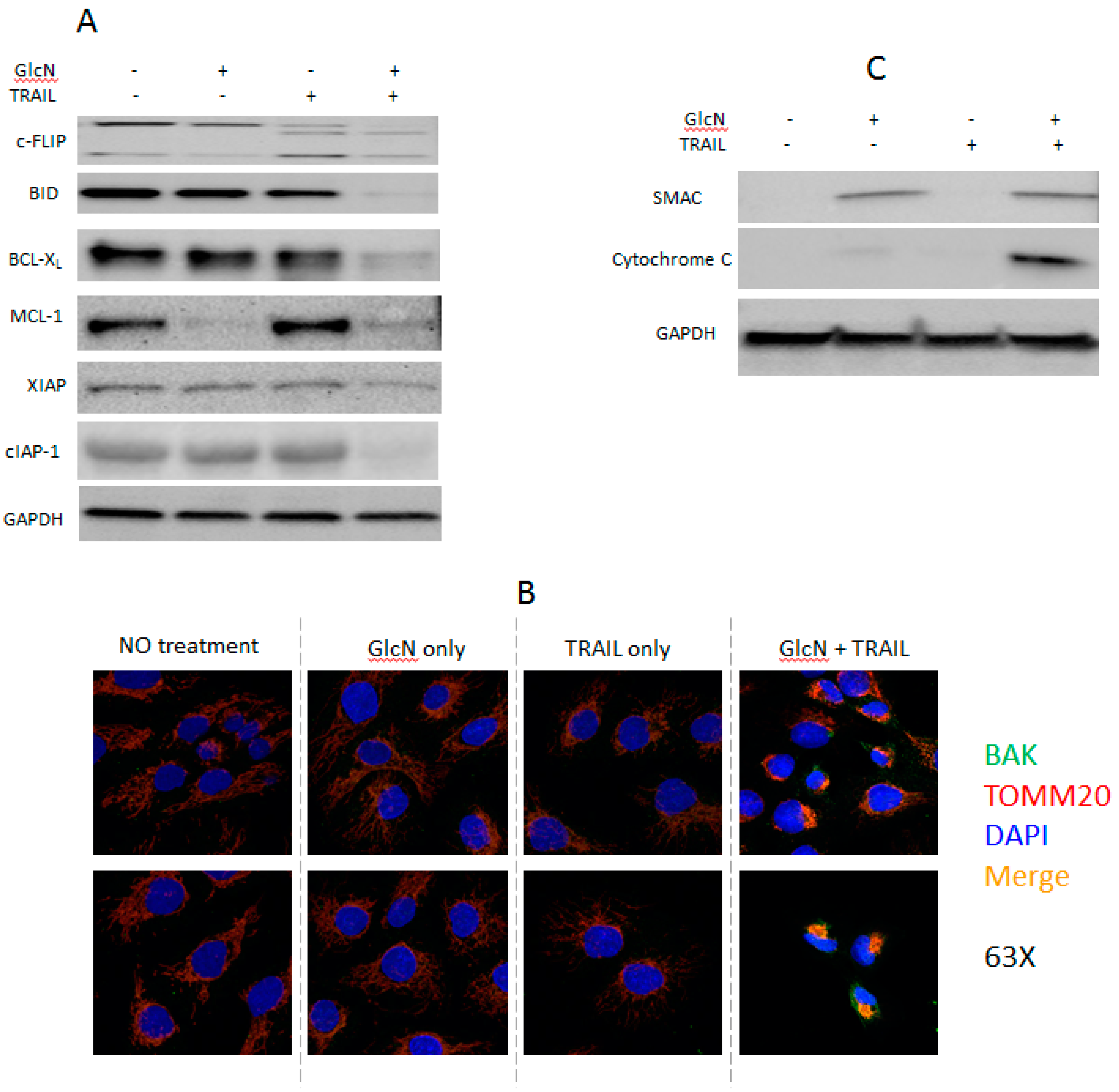
3.4. GlcN and TRAIL Treatment Overcome TRAIL-Resistance through Multiple Pathways
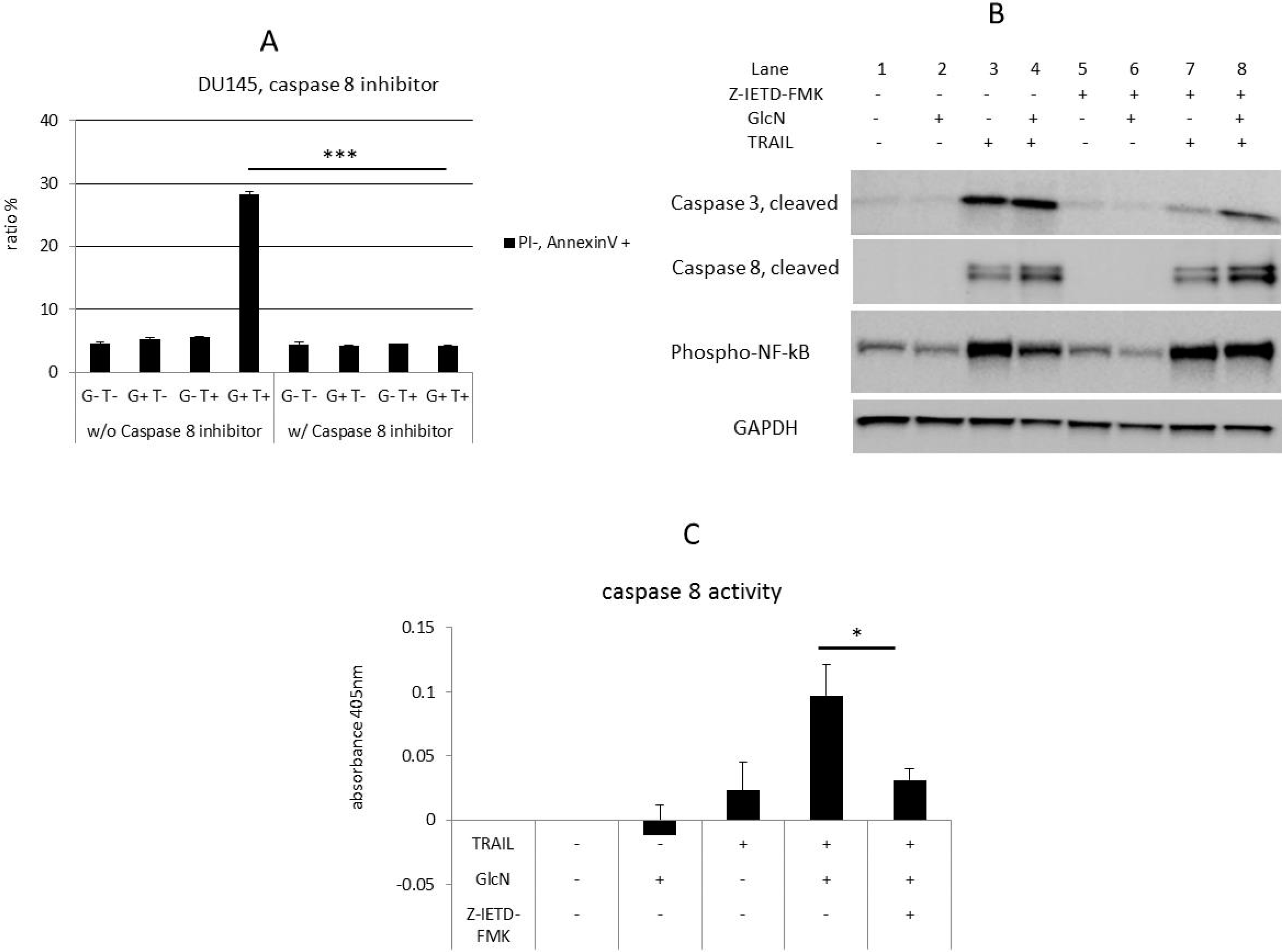
3.5. GlcN Suppresses TRAIL-Activated NF-kB in DU145 Cells
3.6. GlcN Enhanced TRAIL-Induced Apoptosis is Caspase 8-Dependent
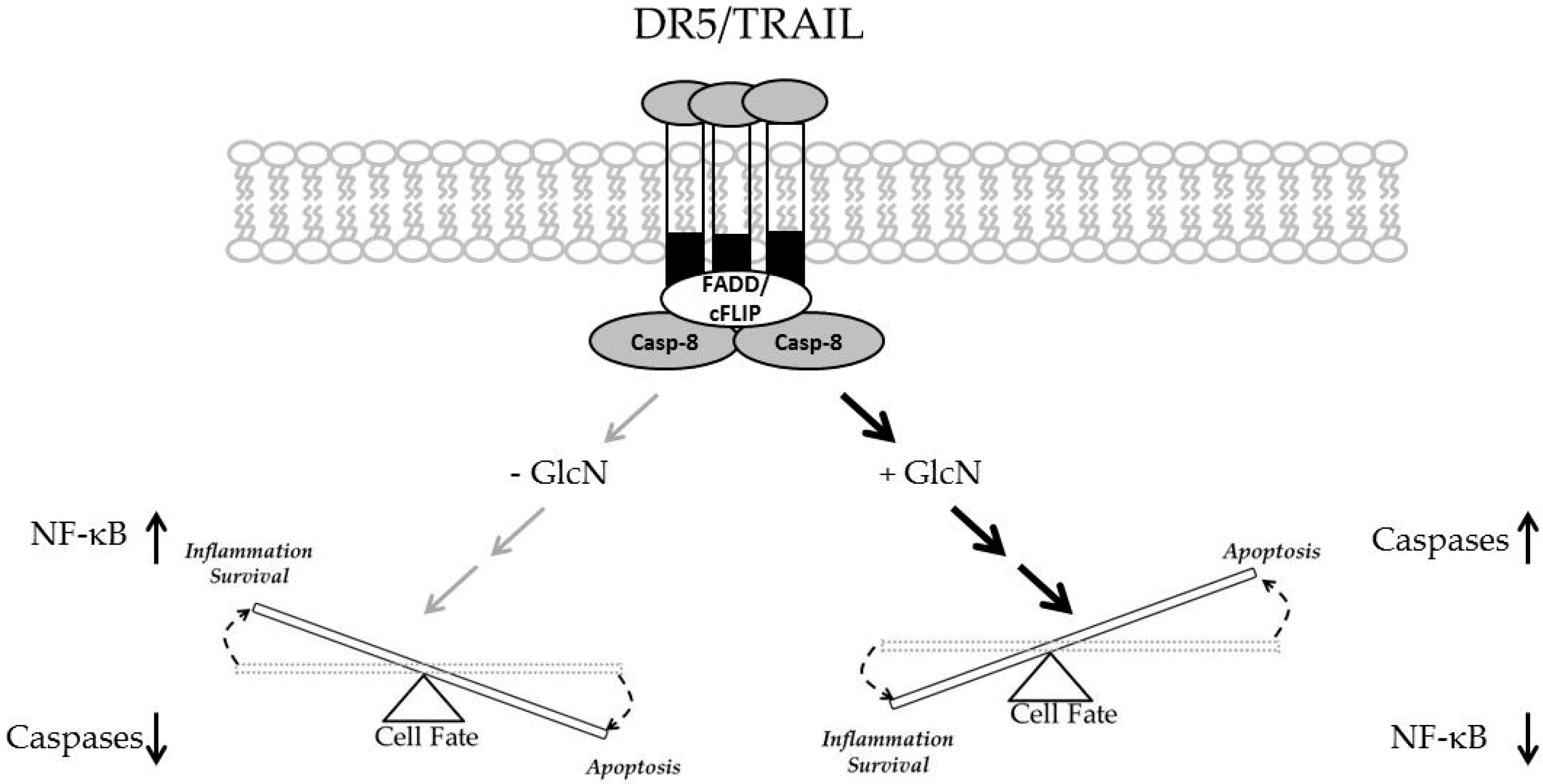
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Voortman, J.; Resende, T.P.; Abou El Hassan, M.A.; Giaccone, G.; Kruyt, F.A. TRAIL therapy in non-small cell lung cancer cells: Sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Mol. Cancer Ther. 2007, 6, 2103–2112. [Google Scholar] [CrossRef]
- van Geelen, C.M.; de Vries, E.G.; de Jong, S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: Paving the road to patient-tailored therapy. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2004, 7, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, A.; Lu, X.; Johnson, M.; Miller, G.J.; Ivashchenko, Y.; Kraft, A.S. Elevated AKT activity protects the prostate cancer cell line LNCaP from TRAIL-induced apoptosis. J. Biol. Chem. 2001, 276, 10767–10774. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Marzo, I.; Anel, A.; Naval, J. Targeting the Apo2L/TRAIL system for the therapy of autoimmune diseases and cancer. Biochem. Pharmacol. 2012, 83, 1475–1483. [Google Scholar] [CrossRef]
- Refaat, A.; Abd-Rabou, A.; Reda, A. TRAIL combinations: The new ‘trail’ for cancer therapy (Review). Oncol. Lett. 2014, 7, 1327–1332. [Google Scholar] [CrossRef]
- Nagane, M.; Pan, G.; Weddle, J.J.; Dixit, V.M.; Cavenee, W.K.; Huang, H.J. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 2000, 60, 847–853. [Google Scholar]
- Shiraishi, T.; Yoshida, T.; Nakata, S.; Horinaka, M.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005, 65, 6364–6370. [Google Scholar] [CrossRef]
- Jung, Y.H.; Lim, E.J.; Heo, J.; Kwon, T.K.; Kim, Y.H. Tunicamycin sensitizes human prostate cells to TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of cIAP2. Int. J. Oncol. 2012, 40, 1941–1948. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anti-Cancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Yue, P.; Khuri, F.R.; Sun, S.Y. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008, 68, 7484–7492. [Google Scholar] [CrossRef] [PubMed]
- Elbein, A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit. Rev. Biochem. 1984, 16, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Bourke, C.A.; Carrigan, M.J. Experimental tunicamycin toxicity in cattle, sheep and pigs. Aust. Vet. J. 1993, 70, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.U.; Schwarz, R.T.; Scholtissek, C. Glucosamine itself mediates reversible inhibition of protein glycosylation. A study of glucosamine metabolism at inhibitory concentrations in influenza-virus-infected cells. Eur. J. Biochem. 1979, 94, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. 2), 24S–42S. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Yerbes, R.; Lopez-Rivas, A.; Reginato, M.J.; Palacios, C. Control of FLIP(L) expression and TRAIL resistance by the extracellular signal-regulated kinase1/2 pathway in breast epithelial cells. Cell Death Differ. 2012, 19, 1908–1916. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Saraei, R.; Soleimani, M.; Movassaghpour Akbari, A.A.; Farshdousti Hagh, M.; Hassanzadeh, A.; Solali, S. The role of XIAP in resistance to TNF-related apoptosis-inducing ligand (TRAIL) in Leukemia. Biomed. Pharmacother. 2018, 107, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dittmer, M.R.; Blackwell, K.; Workman, L.M.; Hostager, B.; Habelhah, H. TRAIL activates JNK and NF-kappaB through RIP1-dependent and -independent pathways. Cell. Signal. 2015, 27, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, H.; Fulda, S.; Schmid, I.; Hiscott, J.; Debatin, K.M.; Jeremias, I. TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene 2003, 22, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. Int. J. Pathol. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Szliszka, E.; Helewski, K.J.; Mizgala, E.; Krol, W. The dietary flavonol fisetin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells. Int. J. Oncol. 2011, 39, 771–779. [Google Scholar] [CrossRef]
- Labsch, S.; Liu, L.; Bauer, N.; Zhang, Y.; Aleksandrowicz, E.; Gladkich, J.; Schonsiegel, F.; Herr, I. Sulforaphane and TRAIL induce a synergistic elimination of advanced prostate cancer stem-like cells. Int. J. Oncol. 2014, 44, 1470–1480. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Waterhouse, N.J.; Steel, R.; Kluck, R.; Trapani, J.A. Assaying cytochrome C translocation during apoptosis. Methods Mol. Biol. 2004, 284, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J. Endoplasmic reticulum stress: A key player in human disease. FEBS J. 2019, 286, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Chesnokov, V.; Gong, B.; Sun, C.; Itakura, K. Anti-cancer activity of glucosamine through inhibition of N-linked glycosylation. Cancer Cell Int. 2014, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Edagawa, M.; Kawauchi, J.; Hirata, M.; Goshima, H.; Inoue, M.; Okamoto, T.; Murakami, A.; Maehara, Y.; Kitajima, S. Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J. Biol. Chem. 2014, 289, 21544–21561. [Google Scholar] [CrossRef] [PubMed]
- Beriault, D.R.; Werstuck, G.H. The role of glucosamine-induced ER stress in diabetic atherogenesis. Exp. Diabetes Res. 2012, 2012, 187018. [Google Scholar] [CrossRef]
- Yoshida, T.; Shiraishi, T.; Horinaka, M.; Wakada, M.; Sakai, T. Glycosylation modulates TRAIL-R1/death receptor 4 protein: Different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 2007, 18, 1239–1242. [Google Scholar] [CrossRef]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Jiang, C.; Schuster, T.; Li, G.X.; Daniel, P.T.; Lu, J. Inorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax-mediated activation of mitochondrial pathway. Mol. Cancer Ther. 2006, 5, 1873–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis Int. J. Program. Cell Death 2017, 22, 898–919. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Middleton, J.; Kim, T.; Lagana, A.; Piovan, C.; Secchiero, P.; Nuovo, G.J.; Cui, R.; Joshi, P.; Romano, G.; et al. A set of NF-kappaB-regulated microRNAs induces acquired TRAIL resistance in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E3355–E3364. [Google Scholar] [CrossRef] [PubMed]
- Lirdprapamongkol, K.; Sakurai, H.; Suzuki, S.; Koizumi, K.; Prangsaengtong, O.; Viriyaroj, A.; Ruchirawat, S.; Svasti, J.; Saiki, I. Vanillin enhances TRAIL-induced apoptosis in cancer cells through inhibition of NF-kappaB activation. In Vivo 2010, 24, 501–506. [Google Scholar]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Ashkenazi, A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Yadav, V.R.; Ravindran, J.; Aggarwal, B.B. RETRACTED**ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2011, 71, 538–549. [Google Scholar] [CrossRef]
- He, Q.; Huang, Y.; Sheikh, M.S. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene 2004, 23, 2554–2558. [Google Scholar] [CrossRef]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.H.; Jiang, C.C.; Kiejda, K.A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis 2007, 28, 2328–2336. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Jiang, C.C.; Lavis, C.J.; Croft, A.; Dong, L.; Tseng, H.Y.; Yang, F.; Tay, K.H.; Hersey, P.; Zhang, X.D. 2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2. Mol. Cancer 2009, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G.; Li, L.; Chopra, D.P.; Porter, A.T. Extended survivability of prostate cancer cells in the absence of trophic factors: Increased proliferation, evasion of apoptosis, and the role of apoptosis proteins. Cancer Res. 1998, 58, 3466–3479. [Google Scholar] [PubMed]
- Liang, Y.; Xu, W.; Liu, S.; Chi, J.; Zhang, J.; Sui, A.; Wang, L.; Liang, Z.; Li, D.; Chen, Y.; et al. N-Acetyl-Glucosamine Sensitizes Non-Small Cell Lung Cancer Cells to TRAIL-Induced Apoptosis by Activating Death Receptor 5. Cell. Physiol. Biochem. 2018, 45, 2054–2070. [Google Scholar] [CrossRef] [PubMed]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef]
- Borrelli, S.; Candi, E.; Alotto, D.; Castagnoli, C.; Melino, G.; Vigano, M.A.; Mantovani, R. p63 regulates the caspase-8-FLIP apoptotic pathway in epidermis. Cell Death Differ. 2009, 16, 253–263. [Google Scholar] [CrossRef]
- Golks, A.; Brenner, D.; Fritsch, C.; Krammer, P.H.; Lavrik, I.N. c-FLIPR, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 2005, 280, 14507–14513. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Hegde, R.; Saleh, A.; Datta, P.; Shiozaki, E.; Chai, J.; Lee, R.A.; Robbins, P.D.; Fernandes-Alnemri, T.; Shi, Y.; et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001, 410, 112–116. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Chesnokov, V.; Larson, G.; Itakura, K. Glucosamine Enhances TRAIL-Induced Apoptosis in the Prostate Cancer Cell Line DU145. Medicines 2019, 6, 104. https://doi.org/10.3390/medicines6040104
Sun C, Chesnokov V, Larson G, Itakura K. Glucosamine Enhances TRAIL-Induced Apoptosis in the Prostate Cancer Cell Line DU145. Medicines. 2019; 6(4):104. https://doi.org/10.3390/medicines6040104
Chicago/Turabian StyleSun, Chao, Viktor Chesnokov, Garrett Larson, and Keiichi Itakura. 2019. "Glucosamine Enhances TRAIL-Induced Apoptosis in the Prostate Cancer Cell Line DU145" Medicines 6, no. 4: 104. https://doi.org/10.3390/medicines6040104
APA StyleSun, C., Chesnokov, V., Larson, G., & Itakura, K. (2019). Glucosamine Enhances TRAIL-Induced Apoptosis in the Prostate Cancer Cell Line DU145. Medicines, 6(4), 104. https://doi.org/10.3390/medicines6040104