Rare Neurologic Disease-Associated Mutations of AIMP1 Are Related with Inhibitory Neuronal Differentiation Which Is Reversed by Ibuprofen

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Primary and Secondary Antibodies
2.2. Plasmid Constructions
2.3. Cell Culture, Differentiation, Plasmid Transfection, and Isolation of Stable Clones
2.4. Fluorescence Images
2.5. Non-Denatured and Denatured Polyacrylamide Gel Electrophoresis and Immunoblotting
2.6. Immunoprecipitation for the Purposed Protein or the Lysosome
2.7. Mass Spectrometry of an Interacting Protein
2.8. Statistical Analysis
2.9. Ethics Statement
3. Results
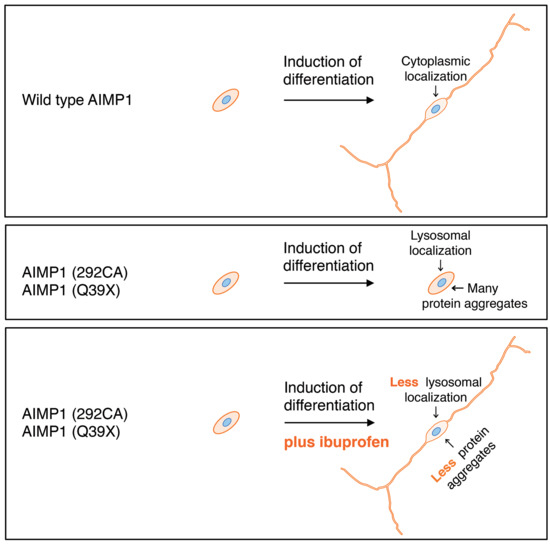
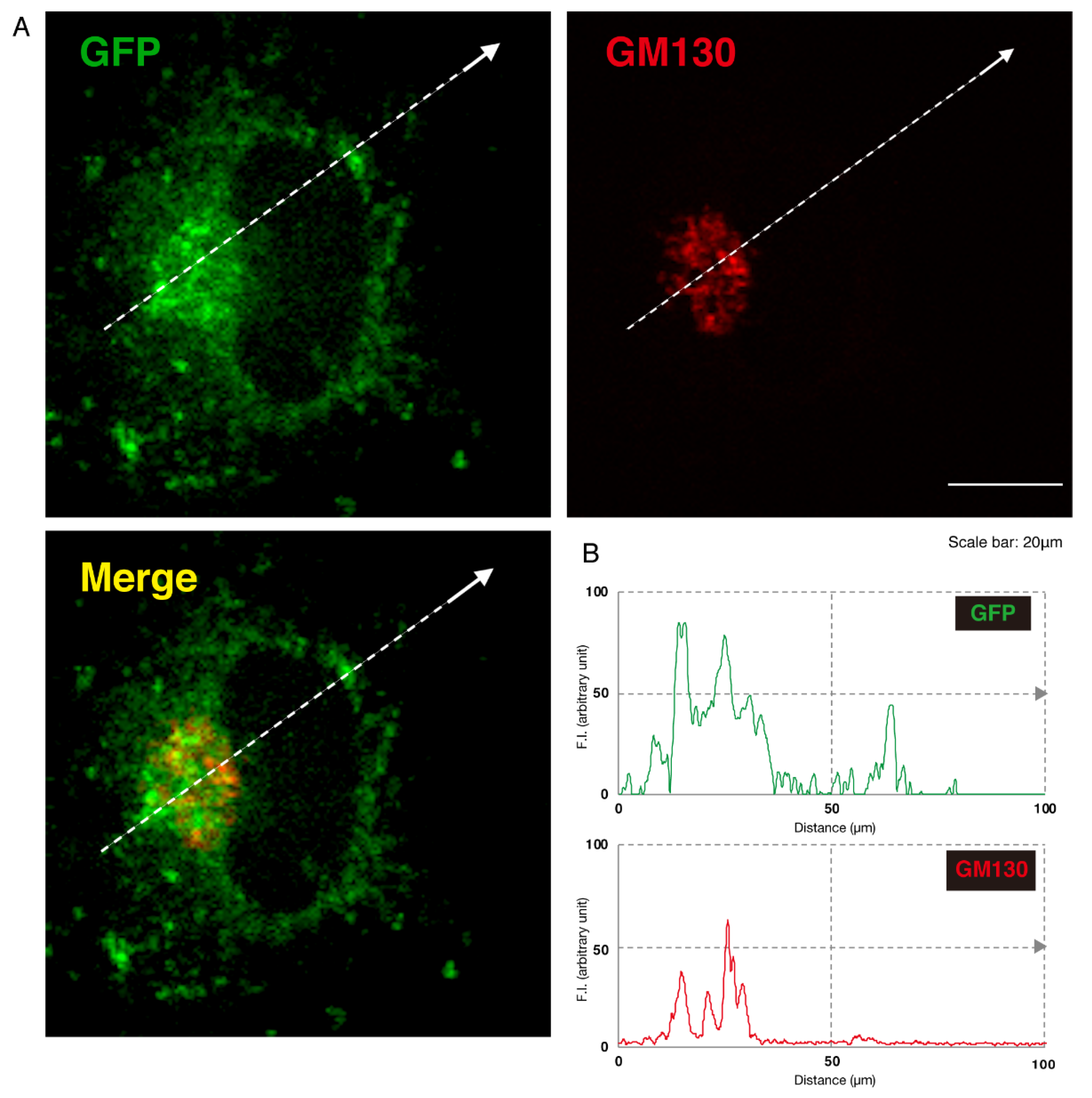
3.1. 292CA or Q39X Mutant Proteins of AIMP1 are Mainly Localized in the Lysosome
3.2. 292CA or Q39X Mutant Proteins Forms Oligomers
3.3. 292CA Mutant Proteins Specifically Interact With Actin
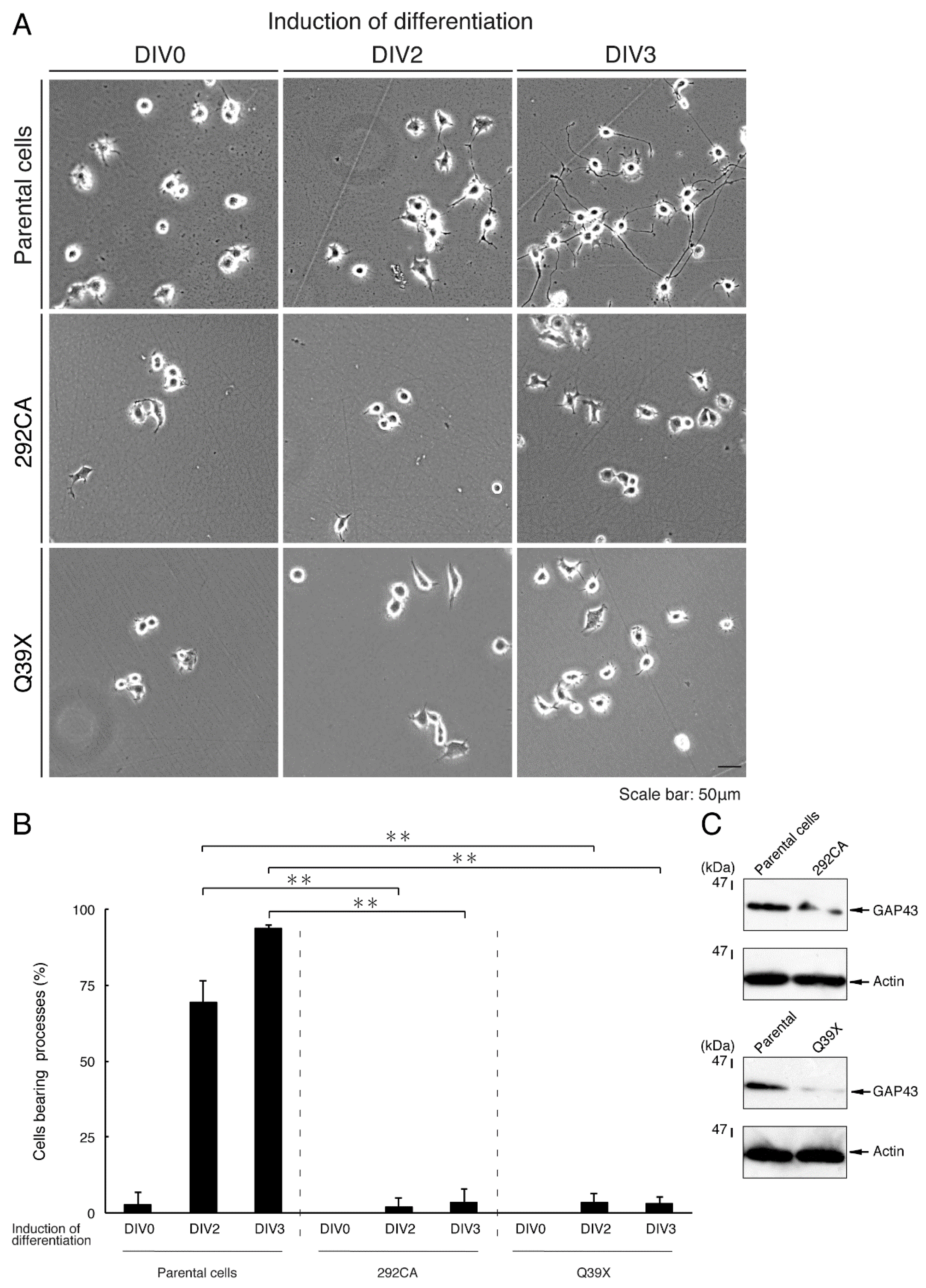
3.4. Cells Harboring the 292CA or Q39X Mutant Constructs Fail to Exhibit Differentiated Phenotypes
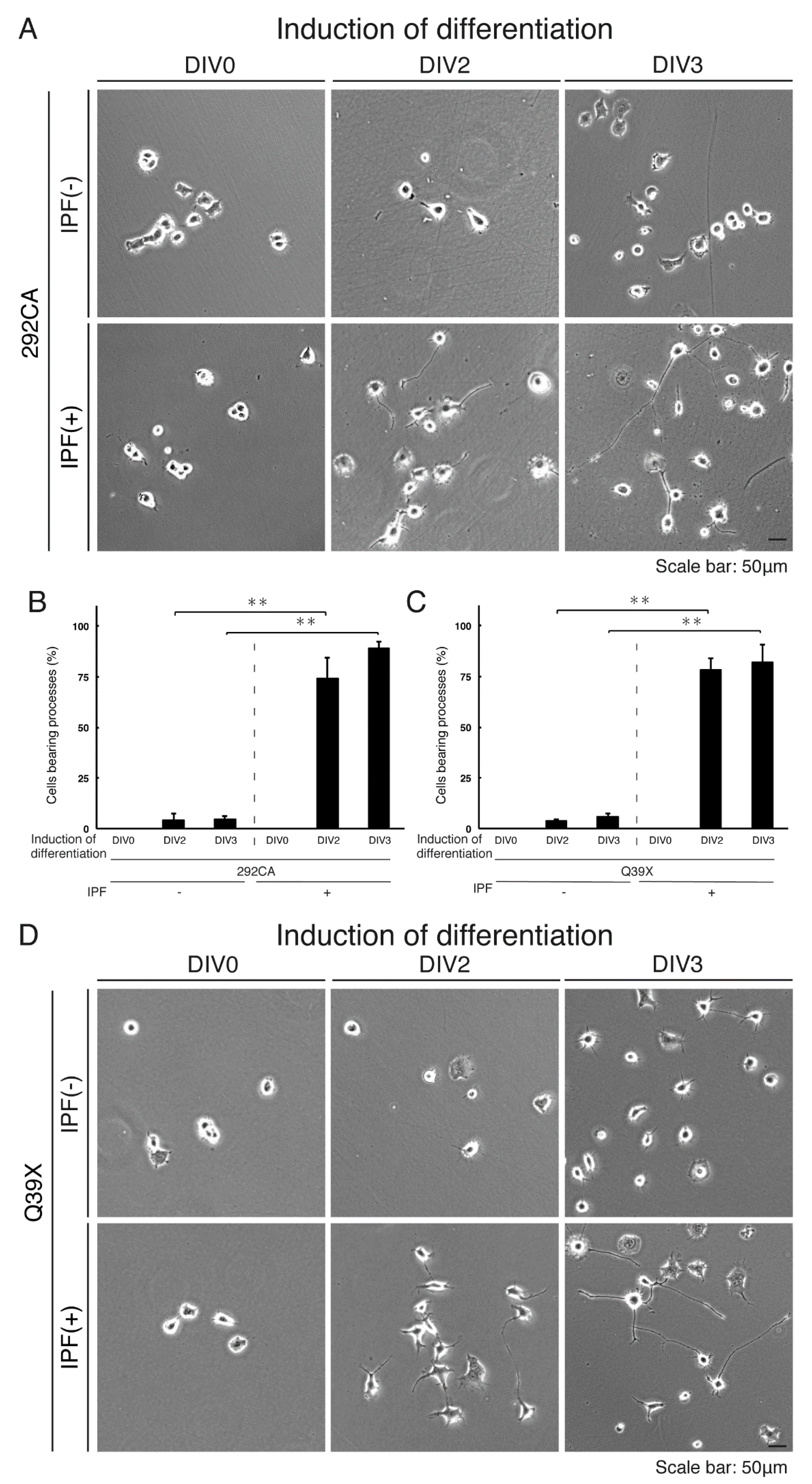
3.5. Ibuprofen Reverses Inhibitory Differentiation of Phenotypes Among Cells Harboring the 292CA or Q39X Mutant Constructs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simons, M.; Lyons, D.A. Axonal selection and myelin sheath generation in the central nervous system. Curr. Opin. Cell Biol. 2013, 25, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Morton, P.D.; Ishibashi, N.; Jonas, R.A.; Gallo, V. Congenital cardiac anomalies and white matter injury. Trends Neurosci. 2015, 38, 353–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rub, M.; Miller, R.H. Emerging cellular and molecular strategies for enhancing central nervous system (CNS) remyelination. Brain Sci. 2018, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Garbern, J.; Cambi, F.; Shy, M.; Kamholz, J. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch. Neurol. 1999, 56, 1210–1214. [Google Scholar] [CrossRef]
- Dhaunchak, A.S.; Colman, D.R.; Nave, K.A. Misalignment of PLP/DM20 transmembrane domains determines protein misfolding in Pelizaeus-Merzbacher disease. J. Neurosci. 2011, 31, 14961–14971. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Lin, Y.; Li, J.; Fenstermaker, A.G.; Way, S.W.; Clayton, B.; Jamison, S.; Harding, H.P.; Ron, D.; Popko, B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 2013, 33, 5980–5991. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K. Cellular pathology of Pelizaeus-Merzbacher disease involving chaperones associated with endoplasmic reticulum stress. Front. Mol. Biosci. 2017, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Biancheri, R.; Rosano, C.; Denegri, L.; Lamantea, E.; Pinto, F.; Lanza, F.; Severino, M.; Filocamo, M. Expanded spectrum of Pelizaeus-Merzbacher-like disease: Literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur. J. Hum. Genet. 2013, 21, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, M.; Markus, B.; Noyman, I.; Shalev, H.; Flusser, H.; Shelef, I.; Liani-Leibson, K.; Shorer, Z.; Cohen, I.; Khateeb, S.; et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010, 87, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Han, J.M.; Kim, S. Protein-protein interactions and multi-component complexes of aminoacyl-tRNA synthetases. Top. Curr. Chem. 2014, 344, 119–144. [Google Scholar] [PubMed]
- Biancheri, R.; Rossi, A.; Zara, F.; Filocamo, M. AIMP1/p43 mutation and PMLD. Am. J. Hum. Genet. 2010, 88, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boespflug-Tanguy, O.; Aubourg, P.; Dorboz, I.; Bégou, M.; Giraud, G.; Sarret, C.; Vaurs-Barrière, C. Neurodegenerative disorder related to AIMP1/p43 mutation is not a PMLD. Am. J. Hum. Genet. 2010, 88, 392–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomason, E.J.; Escalante, M.; Osterhout, D.J.; Fuss, B. The oligodendrocyte growth cone and its actin cytoskeleton: A fundamental element for progenitor cell migration and CNS myelination. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Urai, Y.; Yamawaki, M.; Watanabe, N.; Seki, Y.; Morimoto, T.; Tago, K.; Homma, K.; Sakagami, H.; Miyamoto, Y.; Yamauchi, J. Pull down assay for GTP-bound form of Sar1a reveals its activation during morphological differentiation. Biochem. Biophys. Res. Commun. 2018, 503, 2047–2053. [Google Scholar] [CrossRef]
- Tatsumi, Y.; Matsumoto, N.; Iibe, N.; Watanabe, N.; Torii, T.; Sango, K.; Homma, K.; Miyamoto, Y.; Sakagami, H.; Yamauchi, J. CMT type 2N disease-associated AARS mutant inhibits neurite growth that can be reversed by valproic acid. Neurosci. Res. 2019, 139, 69–78. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 2017, 171, 642–654. [Google Scholar] [CrossRef]
- Avramovich, Y.; Amit, T.; Youdim, M.B. Non-steroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J. Biol. Chem. 2002, 277, 31466–31473. [Google Scholar] [CrossRef] [Green Version]
- Morioka, N.; Kumagai, K.; Morita, K.; Kitayama, S.; Dohi, T. Nonsteroidal anti-inflammatory drugs potentiate 1-methyl-4-phenylpyridinium (MPP+)-induced cell death by promoting the intracellular accumulation of MPP+ in PC12 cells. J. Pharmacol. Exp. Ther. 2004, 310, 800–807. [Google Scholar] [CrossRef]
- Fu, Q.; Hue, J.; Li, S. Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J. Neurosci. 2007, 27, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Zhang, Y.; Oh, M.S.; Schauder, C.M.; Yu, X.; Baskin, J.M.; Dobbs, K.; Notarangelo, L.D.; De Camilli, P.; Walz, T.; et al. Architecture of the human PI4KIII lipid kinase complex. Proc. Natl. Acad. Sci. USA 2017, 114, 13720–13725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zara, F.; Biancheri, R.; Bruno, C.; Bordo, L.; Assereto, S.; Gazzerro, E.; Sotgia, F.; Wang, X.B.; Gianotti, S.; Stringara, S.; et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet. 2006, 38, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Torii, T.; Eguchi, T.; Nakamura, K.; Tanoue, A.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. J. Clin. Neurosci. 2014, 21, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Das Bhowmik, A.; Hebbar, M.; Rajagopal, K.V.; Girisha, K.M.; Gupta, N.; Dalal, A. Homozygosity for a nonsense variant in AIMP2 is associated with a progressive neurodevelopmental disorder with microcephaly, seizures, and spastic quadriparesis. J. Hum. Genet. 2018, 63, 19–25. [Google Scholar] [CrossRef]
- Kwon, H.S.; Park, M.C.; Kim, D.G.; Cho, K.; Park, Y.W.; Han, J.M.; Kim, S. Identification of CD23 as a functional receptor for the proinflammatory cytokine AIMP1/p43. J. Cell Sci. 2012, 125, 4620–4629. [Google Scholar] [CrossRef] [Green Version]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, Y.; Tanaka, M.; Okura, N.; Fukui, Y.; Noguchi, K.; Hayashi, Y.; Torii, T.; Ooizumi, H.; Ohbuchi, K.; Mizoguchi, K.; et al. Rare Neurologic Disease-Associated Mutations of AIMP1 Are Related with Inhibitory Neuronal Differentiation Which Is Reversed by Ibuprofen. Medicines 2020, 7, 25. https://doi.org/10.3390/medicines7050025
Takeuchi Y, Tanaka M, Okura N, Fukui Y, Noguchi K, Hayashi Y, Torii T, Ooizumi H, Ohbuchi K, Mizoguchi K, et al. Rare Neurologic Disease-Associated Mutations of AIMP1 Are Related with Inhibitory Neuronal Differentiation Which Is Reversed by Ibuprofen. Medicines. 2020; 7(5):25. https://doi.org/10.3390/medicines7050025
Chicago/Turabian StyleTakeuchi, Yu, Marina Tanaka, Nanako Okura, Yasuyuki Fukui, Ko Noguchi, Yoshihiro Hayashi, Tomohiro Torii, Hiroaki Ooizumi, Katsuya Ohbuchi, Kazushige Mizoguchi, and et al. 2020. "Rare Neurologic Disease-Associated Mutations of AIMP1 Are Related with Inhibitory Neuronal Differentiation Which Is Reversed by Ibuprofen" Medicines 7, no. 5: 25. https://doi.org/10.3390/medicines7050025
APA StyleTakeuchi, Y., Tanaka, M., Okura, N., Fukui, Y., Noguchi, K., Hayashi, Y., Torii, T., Ooizumi, H., Ohbuchi, K., Mizoguchi, K., Miyamoto, Y., & Yamauchi, J. (2020). Rare Neurologic Disease-Associated Mutations of AIMP1 Are Related with Inhibitory Neuronal Differentiation Which Is Reversed by Ibuprofen. Medicines, 7(5), 25. https://doi.org/10.3390/medicines7050025