

Unique Pulmonary Hypertensive Vascular Diseases Associated with Heart and Lung Developmental Defects

Abstract
:1. Introduction
2. Segmental PH Associated with Complex CHD
2.1. Overview of Segmental PH
2.2. Segmental PH Associated with MAPCAs
2.3. Surgical Treatment for MAPCAs to Avoid Segmental PH
2.4. Pulmonary Vasodilators for Segmental PH
3. Pulmonary Hypertensive Vascular Disease in the Fontan Circulation
3.1. Overview of Fontan Circulation
3.2. Pulmonary Vascular Remodeling in Fontan Circulation
3.3. Pulmonary Vasodilators for Failing Fontan
4. Genetic Origins of PAH Associated with Developmental Defects of the Heart and Lungs
4.1. Overview of Genetics in PAH
4.2. TBX4 in Pediatric PAH
4.3. SOX17 in PAH Associated with CHD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary Arterial Hypertension in France: Results from a National. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Hurdman, J.; Condliffe, R.; Elliot, C.A.; Davies, C.; Hill, C.; Wild, J.M.; Capener, D.; Sephton, P.; Hamilton, N.; Armstrong, I.J.; et al. ASPIRE Registry: Assessing the Spectrum of Pulmonary Hypertension Identified at a REferral Centre. Eur. Respir. J. 2012, 39, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jick, S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary Arterial Hypertension in the USA: An Epidemiological Study in a Large Insured Pediatric Population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, K.; Quarck, R.; Godinas, L.; Belge, C.; Delcroix, M. Learning from Registries in Pulmonary Arterial Hypertension: Pitfalls and Recommendations. Eur. Respir. Rev. 2019, 28, 190050. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric Pulmonary Arterial Hypertension: Updates on Definition, Classification, Diagnostics and Management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Diller, G.; Opotowsky, A.R.; D’Alto, M.; Gu, H.; Giannakoulas, G.; Budts, W.; Broberg, C.S.; Veldtman, G.; Swan, L.; et al. Definition and Management of Segmental Pulmonary Hypertension. J. Am. Heart Assoc. 2018, 7, e008587. [Google Scholar] [CrossRef] [Green Version]
- Alex, A.; Ayyappan, A.; Valakkada, J.; Kramadhari, H.; Sasikumar, D.; Menon, S. Major Aortopulmonary Collateral Arteries. Radiol. Cardiothorac. Imaging 2022, 4, e210157. [Google Scholar] [CrossRef]
- Yasuhara, J.; Yamagishi, H. Pulmonary Arterial Hypertension Associated with Tetralogy of Fallot. Int. Heart J. 2015, 56, S17–S21. [Google Scholar] [CrossRef] [Green Version]
- Nørgaard, M.A.; Alphonso, N.; Cochrane, A.D.; Menahem, S.; Brizard, C.P.; d’Udekem, Y. Major Aorto-Pulmonary Collateral Arteries of Patients with Pulmonary Atresia and Ventricular Septal Defect Are Dilated Bronchial Arteries. Eur. J. Cardio-Thorac. Surg. 2006, 29, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Ikai, A. Surgical Strategies for Pulmonary Atresia with Ventricular Septal Defect Associated with Major Aortopulmonary Collateral Arteries. Gen. Thorac. Cardiovasc. Surg. 2018, 66, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Frescura, C.; Bini, R.M.; Valente, M.; Gallucci, V. Histology of Pulmonary Arterial Supply in Pulmonary Atresia with Ventricular Septal Defect. Circulation 1979, 60, 1066–1074. [Google Scholar] [CrossRef] [Green Version]
- Soquet, J.; Barron, D.J.; d’Udekem, Y. A Review of the Management of Pulmonary Atresia, Ventricular Septal Defect, and Major Aortopulmonary Collateral Arteries. Ann. Thorac. Surg. 2019, 108, 601–612. [Google Scholar] [CrossRef]
- Yamamura, K.; Nagata, H.; Ikeda, K.; Ihara, K.; Hara, T. Efficacy of Bosentan Therapy for Segmental Pulmonary Artery Hypertension Due to Major Aortopulmonary Collateral Arteries in Children. Int. J. Cardiol. 2012, 161, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.S.; Vettukattill, J.J.; Salmon, A.P.; Veldtman, G.R. Sildenafil Therapy in Complex Pulmonary Atresia with Pulmonary Arterial Hypertension. Int. J. Cardiol. 2008, 129, 339–343. [Google Scholar] [CrossRef]
- Schuuring, M.J.; Bouma, B.J.; Cordina, R.; Gatzoulis, M.A.; Budts, W.; Mullen, M.P.; Vis, J.C.; Celermajer, D.; Mulder, B.J.M. Treatment of Segmental Pulmonary Artery Hypertension in Adults with Congenital Heart Disease. Int. J. Cardiol. 2013, 164, 106–110. [Google Scholar] [CrossRef]
- Apostolopoulou, S.C.; Vagenakis, G.; Rammos, S. Pulmonary Vasodilator Therapy in Tetralogy of Fallot with Pulmonary Atresia and Major Aortopulmonary Collaterals: Case Series and Review of Literature. Cardiol. Young 2017, 27, 1861–1864. [Google Scholar] [CrossRef]
- Kaemmerer, A.-S.; Gorenflo, M.; Huscher, D.; Pittrow, D.; Ewert, P.; Pausch, C.; Delcroix, M.; Ghofrani, H.A.; Hoeper, M.M.; Kozlik-Feldmann, R.; et al. Medical Treatment of Pulmonary Hypertension in Adults with Congenital Heart Disease: Updated and Extended Results from the International COMPERA-CHD Registry. Cardiovasc. Diagn. Ther. 2021, 11, 1255–1268. [Google Scholar] [CrossRef]
- Japanese Association of CHD-PH Registry. Available online: https://jacphr.jp/ (accessed on 15 May 2023).
- Fontan, F.; Baudet, E. Surgical Repair of Tricuspid Atresia. Thorax 1971, 26, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Rychik, J.; Atz, A.M.; Celermajer, D.S.; Deal, B.J.; Gatzoulis, M.A.; Gewillig, M.H.; Hsia, T.-Y.; Hsu, D.T.; Kovacs, A.H.; McCrindle, B.W.; et al. Evaluation and Management of the Child and Adult with Fontan Circulation: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e234–e284. [Google Scholar] [CrossRef] [PubMed]
- Khairy, P.; Fernandes, S.M.; Mayer, J.E.; Triedman, J.K.; Walsh, E.P.; Lock, J.E.; Landzberg, M.J. Long-Term Survival, Modes of Death, and Predictors of Mortality in Patients with Fontan Surgery. Circulation 2008, 117, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Ohuchi, H.; Inai, K.; Nakamura, M.; Park, I.-S.; Watanabe, M.; Hiroshi, O.; Kim, K.-S.; Sakazaki, H.; Waki, K.; Yamagishi, H.; et al. Mode of Death and Predictors of Mortality in Adult Fontan Survivors: A Japanese Multicenter Observational Study. Int. J. Cardiol. 2019, 276, 74–80. [Google Scholar] [CrossRef]
- Gewillig, M.; Brown, S.C. The Fontan Circulation after 45 Years: Update in Physiology. Heart 2016, 102, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Egbe, A.C.; Miranda, W.R.; Anderson, J.H.; Borlaug, B.A. Hemodynamic and Clinical Implications of Impaired Pulmonary Vascular Reserve in the Fontan Circulation. J. Am. Coll. Cardiol. 2020, 76, 2755–2763. [Google Scholar] [CrossRef]
- Krimly, A.; Jain, C.C.; Egbe, A.; Alzahrani, A.; Al Najashi, K.; Albert-Brotons, D.; Veldtman, G.R. The Pulmonary Vascular Bed in Patients with Functionally Univentricular Physiology and a Fontan Circulation. Cardiol. Young 2021, 31, 1241–1250. [Google Scholar] [CrossRef]
- Goldstein, B.H.; Connor, C.E.; Gooding, L.; Rocchini, A.P. Relation of Systemic Venous Return, Pulmonary Vascular Resistance, and Diastolic Dysfunction to Exercise Capacity in Patients with Single Ventricle Receiving Fontan Palliation. Am. J. Cardiol. 2010, 105, 1169–1175. [Google Scholar] [CrossRef]
- Ohuchi, H.; Hayama, Y.; Nakajima, K.; Kurosaki, K.; Shiraishi, I.; Nakai, M. Incidence, Predictors, and Mortality in Patients with Liver Cancer After Fontan Operation. J. Am. Heart Assoc. 2021, 10, e016617. [Google Scholar] [CrossRef]
- De Lange, C.; Möller, T.; Hebelka, H. Fontan-Associated Liver Disease: Diagnosis, Surveillance, and Management. Front. Pediatr. 2023, 11, 1100514. [Google Scholar] [CrossRef]
- Dori, Y.; Smith, C.L. Lymphatic Disorders in Patients with Single Ventricle Heart Disease. Front. Pediatr. 2022, 10, 828107. [Google Scholar] [CrossRef]
- Sharma, S.; Ruebner, R.L.; Furth, S.L.; Dodds, K.M.; Rychik, J.; Goldberg, D.J. Assessment of Kidney Function in Survivors Following Fontan Palliation. Congenit. Heart Dis. 2016, 11, 630–636. [Google Scholar] [CrossRef]
- Diller, G.-P.; Giardini, A.; Dimopoulos, K.; Gargiulo, G.; Muller, J.; Derrick, G.; Giannakoulas, G.; Khambadkone, S.; Lammers, A.E.; Picchio, F.M.; et al. Predictors of Morbidity and Mortality in Contemporary Fontan Patients: Results from a Multicenter Study Including Cardiopulmonary Exercise Testing in 321 Patients. Eur. Heart J. 2010, 31, 3073–3083. [Google Scholar] [CrossRef]
- Kurotobi, S.; Sano, T.; Kogaki, S.; Matsushita, T.; Miwatani, T.; Takeuchi, M.; Matsuda, H.; Okada, S. Bidirectional Cavopulmonary Shunt with Right Ventricular Outflow Patency: The Impact of Pulsatility on Pulmonary Endothelial Function. J. Thorac. Cardiovasc. Surg. 2001, 121, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zongtao, Y.; Huishan, W.; Zengwei, W.; Hongyu, Z.; Minhua, F.; Xinmin, L.; Nanbin, Z.; Hongguang, H. Experimental Study of Nonpulsatile Flow Perfusion and Structural Remodeling of Pulmonary Microcirculation Vessels. Thorac. Cardiovasc. Surg. 2010, 58, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Henaine, R.; Vergnat, M.; Bacha, E.A.; Baudet, B.; Lambert, V.; Belli, E.; Serraf, A. Effects of Lack of Pulsatility on Pulmonary Endothelial Function in the Fontan Circulation. J. Thorac. Cardiovasc. Surg. 2013, 146, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Kalia, K.; Walker-Smith, P.; Ordoñez, M.V.; Barlatay, F.G.; Chen, Q.; Weaver, H.; Caputo, M.; Stoica, S.; Parry, A.; Tulloh, R.M.R. Does Maintenance of Pulmonary Blood Flow Pulsatility at the Time of the Fontan Operation Improve Hemodynamic Outcome in Functionally Univentricular Hearts? Pediatr. Cardiol. 2021, 42, 1180–1189. [Google Scholar] [CrossRef]
- Barton, M.; Yanagisawa, M. Endothelin: 30 Years from Discovery to Therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Imai, Y.; Takanashi, Y.; Seo, K.; Terada, M.; Aoki, M.; Nakazawa, M. Time Course of Endothelin-1 and Adrenomedullin after the Fontan Procedure. Ann. Thorac. Surg. 1999, 68, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Kurosawa, H.; Hashimoto, K.; Nomura, K.; Kitamura, N. The Role of Plasma Endothelin in the Fontan Circulation. J. Cardiovasc. Surg. 2002, 43, 793–797. [Google Scholar]
- Ishida, H.; Kogaki, S.; Ichimori, H.; Narita, J.; Nawa, N.; Ueno, T.; Takahashi, K.; Kayatani, F.; Kishimoto, H.; Nakayama, M.; et al. Overexpression of Endothelin-1 and Endothelin Receptors in the Pulmonary Arteries of Failed Fontan Patients. Int. J. Cardiol. 2012, 159, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Ridderbos, F.-J.S.; Wolff, D.; Timmer, A.; van Melle, J.P.; Ebels, T.; Dickinson, M.G.; Timens, W.; Berger, R.M.F. Adverse Pulmonary Vascular Remodeling in the Fontan Circulation. J. Heart Lung Transplant. 2015, 34, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Kogaki, S.; Takahashi, K.; Ozono, K. Attenuation of Bone Morphogenetic Protein Receptor Type 2 Expression in the Pulmonary Arteries of Patients with Failed Fontan Circulation. J. Thorac. Cardiovasc. Surg. 2012, 143, e24–e26. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-Inducible Factor Signaling in Pulmonary Hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Uebing, A.; Hansen, J.H. Pulmonary Vascular Disease in Fontan Circulation—Is There a Rationale for Pulmonary Vasodilator Therapies? Cardiovasc. Diagn. Ther. 2021, 11, 1111–1121. [Google Scholar] [CrossRef]
- Suda, K.; Matsumura, M.; Miyanish, S.; Uehara, K.; Sugita, T.; Matsumoto, M. Increased Vascular Endothelial Growth Factor in Patients with Cyanotic Congenital Heart Diseases May Not Be Normalized after a Fontan Type Operation. Ann. Thorac. Surg. 2004, 78, 942–946. [Google Scholar] [CrossRef]
- Khambadkone, S.; Li, J.; de Leval, M.R.; Cullen, S.; Deanfield, J.E.; Redington, A.N. Basal Pulmonary Vascular Resistance and Nitric Oxide Responsiveness Late After Fontan-Type Operation. Circulation 2003, 107, 3204–3208. [Google Scholar] [CrossRef] [Green Version]
- Giardini, A.; Balducci, A.; Specchia, S.; Gargiulo, G.; Bonvicini, M.; Picchio, F.M. Effect of Sildenafil on Haemodynamic Response to Exercise and Exercise Capacity in Fontan Patients. Eur. Heart J. 2008, 29, 1681–1687. [Google Scholar] [CrossRef]
- Goldberg, D.J.; French, B.; McBride, M.G.; Marino, B.S.; Mirarchi, N.; Hanna, B.D.; Wernovsky, G.; Paridon, S.M.; Rychik, J. Impact of Oral Sildenafil on Exercise Performance in Children and Young Adults After the Fontan Operation. Circulation 2011, 123, 1185–1193. [Google Scholar] [CrossRef]
- Collins, J.L.G.; Law, M.A.; Borasino, S.; Erwin, W.C.; Cleveland, D.C.; Alten, J.A. Routine Sildenafil Does Not Improve Clinical Outcomes After Fontan Operation. Pediatr. Cardiol. 2017, 38, 1703–1708. [Google Scholar] [CrossRef]
- Goldberg, D.J.; Zak, V.; Goldstein, B.H.; Schumacher, K.R.; Rhodes, J.; Penny, D.J.; Petit, C.J.; Ginde, S.; Menon, S.C.; Kim, S.-H.; et al. Results of the FUEL Trial. Circulation 2020, 141, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Schuuring, M.J.; Vis, J.C.; van Dijk, A.P.J.; van Melle, J.P.; Vliegen, H.W.; Pieper, P.G.; Sieswerda, G.T.; de Bruin-Bon, R.H.A.C.M.; Mulder, B.J.M.; Bouma, B.J. Impact of Bosentan on Exercise Capacity in Adults after the Fontan Procedure: A Randomized Controlled Trial. Eur. J. Heart Fail. 2013, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Li, Y.; Liu, M.; Zhou, H.; Peng, T.; Deng, X.; Tao, L.; Zhang, G. Efficacy of Endothelin Receptor Antagonist Bosentan on the Long-Term Prognosis in Patients after Fontan Operation. Zhonghua Xin Xue Guan Bing Za Zhi 2013, 41, 1025–1028. [Google Scholar]
- Cedars, A.M.; Saef, J.; Peterson, L.R.; Coggan, A.R.; Novak, E.L.; Kemp, D.; Ludbrook, P.A. Effect of Ambrisentan on Exercise Capacity in Adult Patients After the Fontan Procedure. Am. J. Cardiol. 2016, 117, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Ubeda-Tikkanen, A.; Clair, M.; Fernandes, S.M.; Graham, D.A.; Milliren, C.E.; Daly, K.P.; Mullen, M.P.; Landzberg, M.J. Effect of Inhaled Iloprost on the Exercise Function of Fontan Patients: A Demonstration of Concept. Int. J. Cardiol. 2013, 168, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hu, X.; Liao, W.; Rutahoile, W.H.; Malenka, D.J.; Zeng, X.; Yang, Y.; Feng, P.; Wen, L.; Huang, W. The Efficacy and Safety of Pulmonary Vasodilators in Patients with Fontan Circulation: A Meta-analysis of Randomized Controlled Trials. Pulm. Circ. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhou, X.; An, Q.; Feng, Y. Pulmonary Vasodilator Therapy after the Fontan Procedure: A Meta-Analysis. Heart Fail. Rev. 2021, 26, 91–100. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and Genomics of Pulmonary Arterial Hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel Risk Genes and Mechanisms Implicated by Exome Sequencing of 2572 Individuals with Pulmonary Arterial Hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Eichstaedt, C.A.; Saßmannshausen, Z.; Shaukat, M.; Cao, D.; Xanthouli, P.; Gall, H.; Sommer, N.; Ghofrani, H.-A.; Seyfarth, H.-J.; Lerche, M.; et al. Gene Panel Diagnostics Reveals New Pathogenic Variants in Pulmonary Arterial Hypertension. Respir. Res. 2022, 23, 74. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of Rare Sequence Variation Underlying Heritable Pulmonary Arterial Hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [Green Version]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular Genetic Framework Underlying Pulmonary Arterial Hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous Germline Mutations in BMPR2, Encoding a TGF-β Receptor, Cause Familial Primary Pulmonary Hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A.; Hamid, R.; Loyd, J.E. Longitudinal Analysis Casts Doubt on the Presence of Genetic Anticipation in Heritable Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 Mutations and Survival in Pulmonary Arterial Hypertension: An Individual Participant Data Meta-Analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zeng, Q.; Ma, Y.; Liu, B.; Chen, Q.; Li, W.; Xiong, C.; Zhou, Z. Genetic Analyses in a Cohort of 191 Pulmonary Arterial Hypertension Patients. Respir. Res. 2018, 19, 87. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jaïs, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical Outcomes of Pulmonary Arterial Hypertension in Patients Carrying an ACVRL1 (ALK1) Mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Lian, T.-Y.; Jiang, X.; Liu, S.-F.; Li, S.-Q.; Jiang, R.; Wu, W.-H.; Ye, J.; Cheng, C.-Y.; Du, Y.; et al. Germline BMP9 Mutation Causes Idiopathic Pulmonary Arterial Hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Yoshida, Y.; Uchida, K.; Kodo, K.; Shibata, H.; Furutani, Y.; Nakayama, T.; Sakai, S.; Nakanishi, T.; Takahashi, T.; Yamagishi, H. Genetic and Functional Analyses of TBX4 Reveal Novel Mechanisms Underlying Pulmonary Arterial Hypertension. J. Mol. Cell Cardiol. 2022, 171, 105–116. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.H.F.; Roofthooft, M.T.R.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 Mutations (Small Patella Syndrome) Are Associated with Childhood-Onset Pulmonary Arterial Hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Yoshida, Y.; Kodo, K.; Yamagishi, H. Roles of Tbx4 in the lung mesenchyme for airway and vascular development. In Molecular Mechanism of Congenital Heart Disease and Pulmonary Hypertension; Nakanishi, T., Baldwin, H.S., Fineman, J.R., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2020; pp. 79–81. [Google Scholar]
- Sheeba, C.J.; Logan, M.P.O. The roles of T-box genes in vertebrate limb development. Curr. Top. Dev. Biol. 2017, 122, 355–381. [Google Scholar]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic Analyses in a Cohort of Children with Pulmonary Hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome Sequencing in Children with Pulmonary Arterial Hypertension Demonstrates Differences Compared with Adults. Circ. Genom. Precis. Med. 2018, 11, e001887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galambos, C.; Mullen, M.P.; Shieh, J.T.; Schwerk, N.; Kielt, M.J.; Ullmann, N.; Boldrini, R.; Stucin-Gantar, I.; Haass, C.; Bansal, M.; et al. Phenotype Characterisation of TBX4 Mutation and Deletion Carriers with Neonatal and Paediatric Pulmonary Hypertension. Eur. Respir. J. 2019, 54, 1801965. [Google Scholar] [CrossRef]
- Szafranski, P.; Coban-Akdemir, Z.H.; Rupps, R.; Grazioli, S.; Wensley, D.; Jhangiani, S.N.; Popek, E.; Lee, A.F.; Lupski, J.R.; Boerkoel, C.F.; et al. Phenotypic Expansion of TBX4 Mutations to Include Acinar Dysplasia of the Lungs. Am. J. Med. Genet. A 2016, 170, 2440–2444. [Google Scholar] [CrossRef]
- German, K.; Deutsch, G.H.; Freed, A.S.; Dipple, K.M.; Chabra, S.; Bennett, J.T. Identification of a Deletion Containing TBX4 in a Neonate with Acinar Dysplasia by Rapid Exome Sequencing. Am. J. Med. Genet. A 2019, 179, 842–845. [Google Scholar] [CrossRef]
- Karolak, J.A.; Vincent, M.; Deutsch, G.; Gambin, T.; Cogné, B.; Pichon, O.; Vetrini, F.; Mefford, H.C.; Dines, J.N.; Golden-Grant, K.; et al. Complex Compound Inheritance of Lethal Lung Developmental Disorders Due to Disruption of the TBX-FGF Pathway. Am. J. Human. Genet. 2019, 104, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Suhrie, K.; Pajor, N.M.; Ahlfeld, S.K.; Dawson, D.B.; Dufendach, K.R.; Kitzmiller, J.A.; Leino, D.; Lombardo, R.C.; Smolarek, T.A.; Rathbun, P.A.; et al. Neonatal Lung Disease Associated with TBX4 Mutations. J. Pediatr. 2019, 206, 286–292.e1. [Google Scholar] [CrossRef] [PubMed]
- Thoré, P.; Girerd, B.; Jaïs, X.; Savale, L.; Ghigna, M.-R.; Eyries, M.; Levy, M.; Ovaert, C.; Servettaz, A.; Guillaumot, A.; et al. Phenotype and Outcome of Pulmonary Arterial Hypertension Patients Carrying a TBX4 Mutation. Eur. Respir. J. 2020, 55, 1902340. [Google Scholar] [CrossRef] [PubMed]
- Naiche, L.A.; Arora, R.; Kania, A.; Lewandoski, M.; Papaioannou, V.E. Identity and Fate of Tbx4-Expressing Cells Reveal Developmental Cell Fate Decisions in the Allantois, Limb, and External Genitalia. Dev. Dyn. 2011, 240, 2290–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, R.; Metzger, R.J.; Papaioannou, V.E. Multiple Roles and Interactions of Tbx4 and Tbx5 in Development of the Respiratory System. PLoS Genet. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Sekine, K.; Ohuchi, H.; Fujiwara, M.; Yamasaki, M.; Yoshizawa, T.; Sato, T.; Yagishita, N.; Matsui, D.; Koga, Y.; Itoh, N.; et al. Fgf10 Is Essential for Limb and Lung Formation. Nat. Genet. 1999, 21, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF10: A Multifunctional Mesenchymal–Epithelial Signaling Growth Factor in Development, Health, and Disease. Cytokine Growth Factor. Rev. 2016, 28, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yan, L.; Kielt, M.J.; Cogan, J.D.; Hedges, L.K.; Nunley, B.; West, J.; Austin, E.D.; Hamid, R. TBX4 Transcription Factor Is a Positive Feedback Regulator of Itself and Phospho-SMAD1/5. Am. J. Respir. Cell Mol. Biol. 2021, 64, 140–143. [Google Scholar] [CrossRef]
- Francois, M.; Koopman, P.; Beltrame, M. SoxF Genes: Key Players in the Development of the Cardio-Vascular System. Int. J. Biochem. Cell Biol. 2010, 42, 445–448. [Google Scholar] [CrossRef]
- Matsui, T.; Kanai-Azuma, M.; Hara, K.; Matoba, S.; Hiramatsu, R.; Kawakami, H.; Kurohmaru, M.; Koopman, P.; Kanai, Y. Redundant Roles of Sox17 and Sox18 in Postnatal Angiogenesis in Mice. J. Cell Sci. 2006, 119, 3513–3526. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Hara, K.; Kanai-Azuma, M.; Matsui, T.; Miura, Y.; Tsunekawa, N.; Kurohmaru, M.; Saijoh, Y.; Koopman, P.; Kanai, Y. Redundant Roles of Sox17 and Sox18 in Early Cardiovascular Development of Mouse Embryos. Biochem. Biophys. Res. Commun. 2007, 360, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lilly, A.J.; Lacaud, G.; Kouskoff, V. SOXF Transcription Factors in Cardiovascular Development. Semin. Cell Dev. Biol. 2017, 63, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Orsenigo, F.; Morini, M.F.; Pitulescu, M.E.; Bhat, G.; Nyqvist, D.; Breviario, F.; Conti, V.; Briot, A.; Iruela-Arispe, M.L.; et al. Sox17 Is Indispensable for Acquisition and Maintenance of Arterial Identity. Nat. Commun. 2013, 4, 2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kim, I.-K.; Ahn, J.S.; Woo, D.-C.; Kim, S.-T.; Song, S.; Koh, G.Y.; Kim, H.-S.; Jeon, B.H.; Kim, I. Deficiency of Endothelium-Specific Transcription Factor Sox17 Induces Intracranial Aneurysm. Circulation 2015, 131, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, A.W.; Haitchi, H.M.; LeCras, T.D.; Sridharan, A.; Xu, Y.; Wert, S.E.; James, J.; Udell, N.; Thurner, P.J.; Whitsett, J.A. Sox17 Is Required for Normal Pulmonary Vascular Morphogenesis. Dev. Biol. 2014, 387, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare Variants in SOX17 Are Associated with Pulmonary Arterial Hypertension with Congenital Heart Disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- McCulley, D.J.; Black, B.L. Transcription Factor Pathways and Congenital Heart Disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar] [PubMed] [Green Version]
- Zorn, A.M.; Barish, G.D.; Williams, B.O.; Lavender, P.; Klymkowsky, M.W.; Varmus, H.E. Regulation of Wnt Signaling by Sox Proteins. Mol. Cell 1999, 4, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Kataoka, M.; Suzuki, H.; Aimi, Y.; Chiba, T.; Kanekura, K.; Satoh, T.; Fukuda, K.; Gamou, S.; Kosaki, K. SOX17 Mutations in Japanese Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 1231–1233. [Google Scholar] [CrossRef] [PubMed]

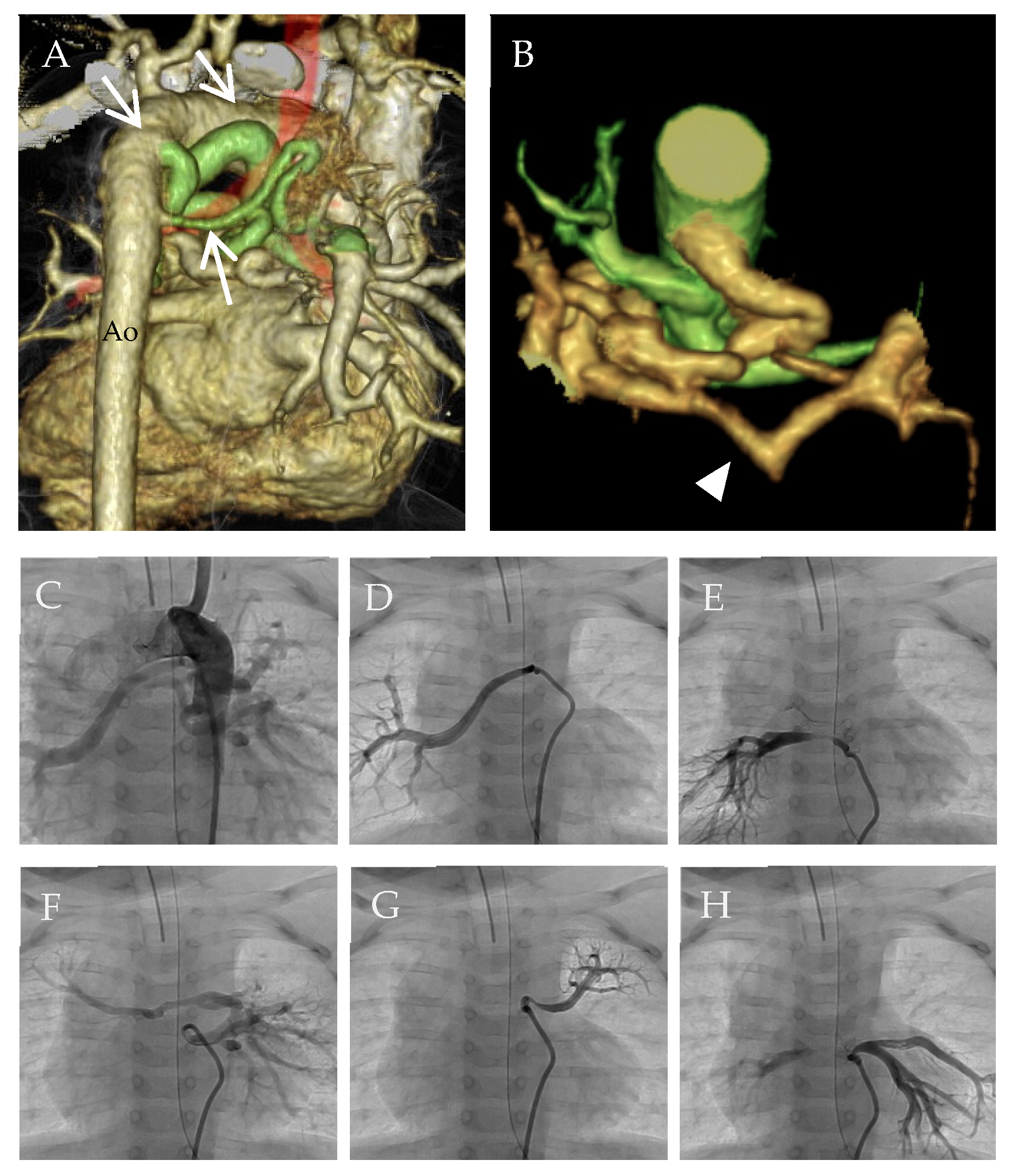
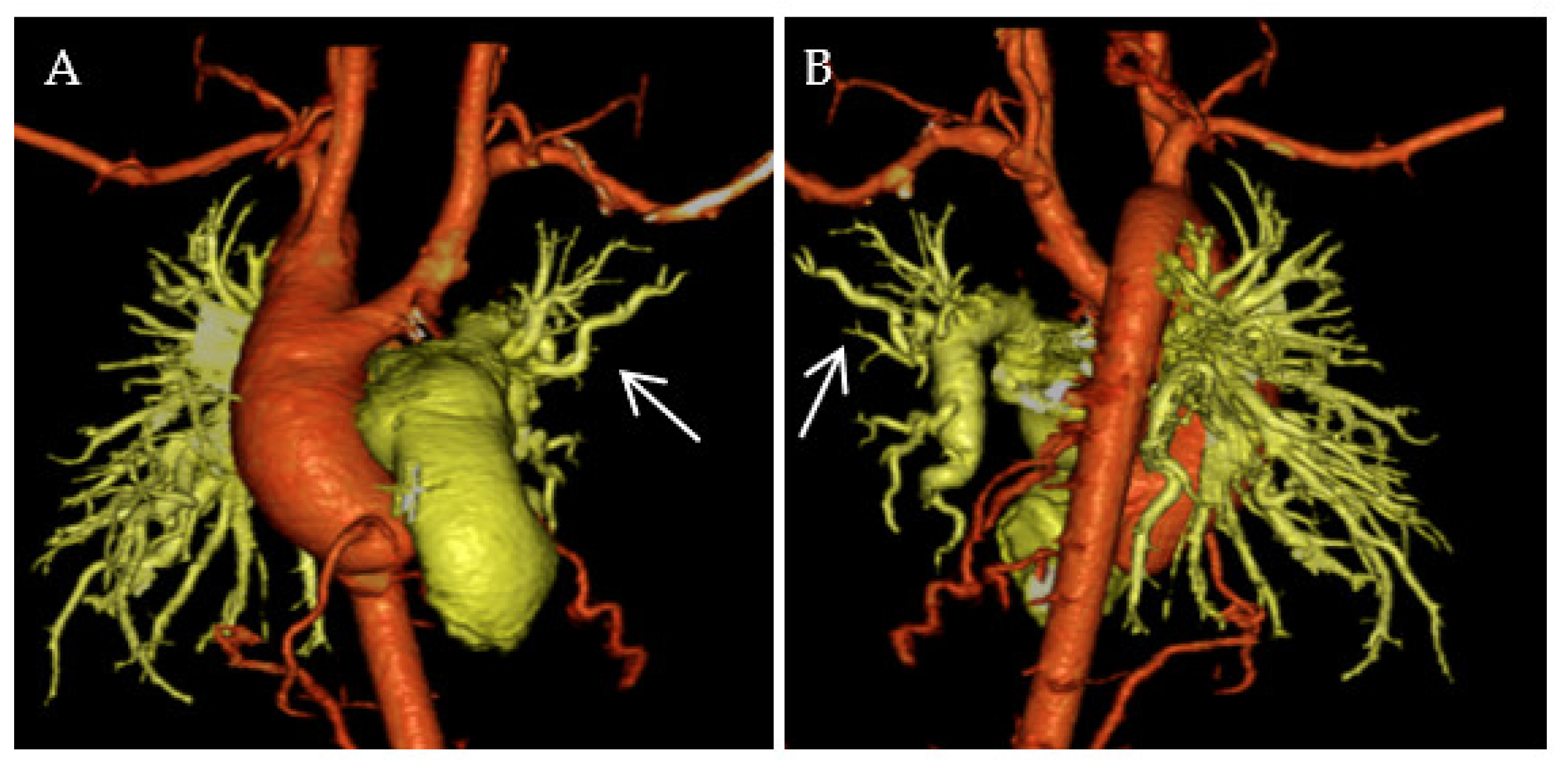

{kind=link}
{kind=link}
{kind=link}
| Group 1 | PAH |
| 1.1 | IPAH |
| 1.2 | HPAH |
| 1.3 | Drug- and toxin-induced PAH |
| 1.4 | APAH |
| 1.4.1 | PAH associated with connective tissue disease (APAH-CTD) |
| 1.4.2 | PAH associated with human immunodeficiency virus infection |
| 1.4.3 | PAH associated with portal hypertension |
| 1.4.4 | APAH-CHD |
| 1.4.5 | PAH associated with schistosomiasis |
| 1.5 | PAH long-term responders to calcium channel blockers |
| 1.6 | PAH with overt features of venous/capillaries (PVOD/PCH) involvement |
| 1.7 | PPHN syndrome |
| Group 2 | PH due to left heart disease |
| 2.1 | PH due to heart failure with preserved left ventricular ejection fraction |
| 2.2 | PH due to heart failure with reduced left ventricular ejection fraction |
| 2.3 | Valvular heart disease |
| 2.4 | Congenital/acquired cardiovascular conditions leading to post-capillary PH |
| Group 3 | PH due to lung diseases and/or hypoxia |
| 3.1 | Obstructive lung disease |
| 3.2 | Restrictive lung disease |
| 3.3 | Other lung disease with mixed restrictive/obstructive pattern |
| 3.4 | Hypoxia without lung disease |
| 3.5 | Developmental lung disorders |
| Group 4 | PH due to pulmonary artery obstructions |
| 4.1 | Chronic thromboembolic pulmonary hypertension |
| 4.2 | Other pulmonary artery obstructions |
| Group 5 | PH with unclear and/or multifactorial mechanisms |
| 5.1 | Hematological disorders |
| 5.2 | Systemic and metabolic disorders |
| 5.3 | Others |
| 5.4 | Complex CHD |
| Study | n | Age (Year) Median (Range) | Duration (Year) Median (Range) | Medication | Outcome |
|---|---|---|---|---|---|
| Lim et al. [17] | 5 | 19 (17–47) | (0.5–2) | S: 5 | Symptoms were improved in 5 cases. 6 MWT was increased in 2 cases. |
| Schuuring et al. [18] | 7 | 32 (23–42) | 1 | B: 7 | Symptoms were improved in 7 cases. 6 MWT was increased in 6 cases. |
| Apostolopoulou et al. [19] | 3 | 26 (21–31) | 10 (5–14) | B 3 | Symptoms were improved in 3 cases. |
| Study | Year | Country | Design | Age | No. of Patients | Drugs | Outcomes | Conclusions |
|---|---|---|---|---|---|---|---|---|
| Giardini et al. [49] | 2008 | Italy | RCT | 22.8 ± 4.9 | 27 | Sildenafil 0.7 mg/kg | peak VO2 | Sildenafil improved exercise capacity, pulmonary blood flow, and cardiac index |
| Goldberg et al. [50] | 2011 | USA | RCT | 14.9 ± 5.1 | 55 | Sildenafil 60 mg/day | peak VO2 | Sildenafil did not improve VO2 max. However, increased ventilatory efficiency and improved oxygen consumption occurred in two subgroups |
| Collins et al. [51] | 2017 | USA | RC | 4.0 ± 0.3 | 103 | Sildenafil 1.05–3.0 mg/kg/day | Chest tube output Length of hospital stay Duration of mechanical ventilation | Routine early administration of sildenafil after Fontan operation did not improve postoperative chest tube output, length of stay, or duration of mechanical ventilation. |
| Goldberg et al. [52] | 2020 | USA | RCT | 15.5 ± 2.0 | 400 | Udenafil 175 mg/day | peak VO2 Anaerobic threshold Myocardial performance index BNP | Udenafil could not improve peak VO2, but improved multiple measures of exercise performance at the anaerobic threshold. |
| Schuuring et al. [53] | 2013 | Netherlands | RCT | 28 (18–55) | 42 | Bosentan 250 mg/day | Exercise capacity NT-proBNP SF-36 quality of life NYHA class | Six months treatment of bosentan was not beneficial. |
| Shang et al. [54] | 2013 | China | RCT | 2.5–18 | 39 | Bosentan, 31.25–250 mg/day according to body weight | Mortality PLE PAF 6MWD NYHA class | Bosentan therapy in post-Fontan patients could reduce the incidence of PAF and PLE and improve heart function. |
| Cedars et al. [55] | 2016 | USA | RCT | 24.9 ± 5.2 | 47 | Ambrisentan 10 mg/day | peak VO2 SF-36 quality of life | Ambrisentan improved exercise capacity in adult Fontan patients. |
| Rhodes et al. [56] | 2013 | USA | RCT | median 16.7 | 18 | Illoprost 5.0 μg | peak VO2 | Iloprost improved the peak oxygen pulse and peak VO2 of Fontan patients and appeared to be particularly beneficial among patients with impaired exercise function. |
| Gene | Pulmonary Hypertension Phenotypic Association | Putative Molecular Mechanism | Inheritance Pattern | Associated Clinical Features | Populations |
|---|---|---|---|---|---|
| TBX4 [62,71,72] | Heritable and idiopathic PAH ischiocoxopodopatellar syndrome Parenchymal lung disease Bronchopulmonary dysplasia Persistent pulmonary hypertension of the neonate | Loss of function | Autosomal dominant | Patellar aplasia Skeletal abnormalities, particularly pelvis, knees, and feet | Pediatric and (less commonly) adult |
| SOX17 [62] | Heritable and idiopathic PAH Congenital heart disease | Unknown | Autosomal dominant | Atrial septal defect, patent ductus arteriosus, and ventricular septal defect | Pediatric and adult |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishida, H.; Maeda, J.; Uchida, K.; Yamagishi, H. Unique Pulmonary Hypertensive Vascular Diseases Associated with Heart and Lung Developmental Defects. J. Cardiovasc. Dev. Dis. 2023, 10, 333. https://doi.org/10.3390/jcdd10080333
Ishida H, Maeda J, Uchida K, Yamagishi H. Unique Pulmonary Hypertensive Vascular Diseases Associated with Heart and Lung Developmental Defects. Journal of Cardiovascular Development and Disease. 2023; 10(8):333. https://doi.org/10.3390/jcdd10080333
Chicago/Turabian StyleIshida, Hidekazu, Jun Maeda, Keiko Uchida, and Hiroyuki Yamagishi. 2023. "Unique Pulmonary Hypertensive Vascular Diseases Associated with Heart and Lung Developmental Defects" Journal of Cardiovascular Development and Disease 10, no. 8: 333. https://doi.org/10.3390/jcdd10080333