
Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target


Abstract
1. Introduction
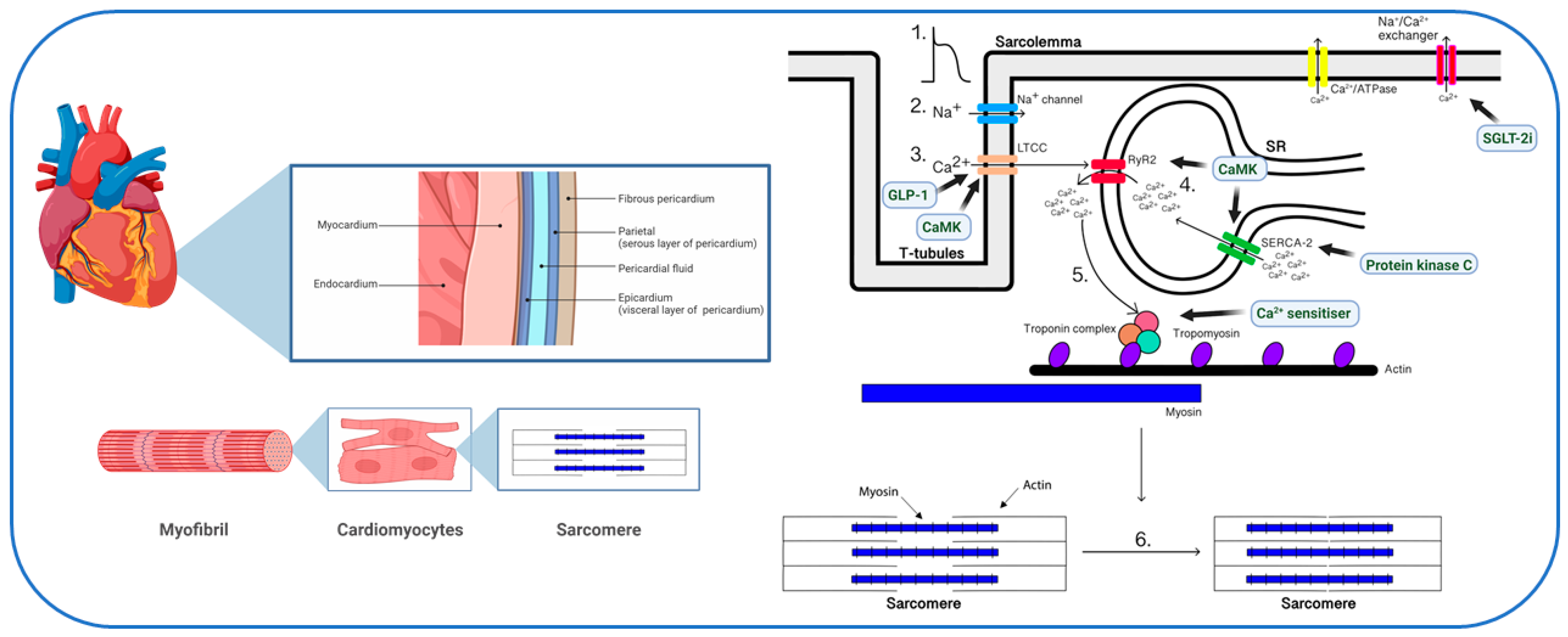
2. Physiology of Myocardial Calcium Handling
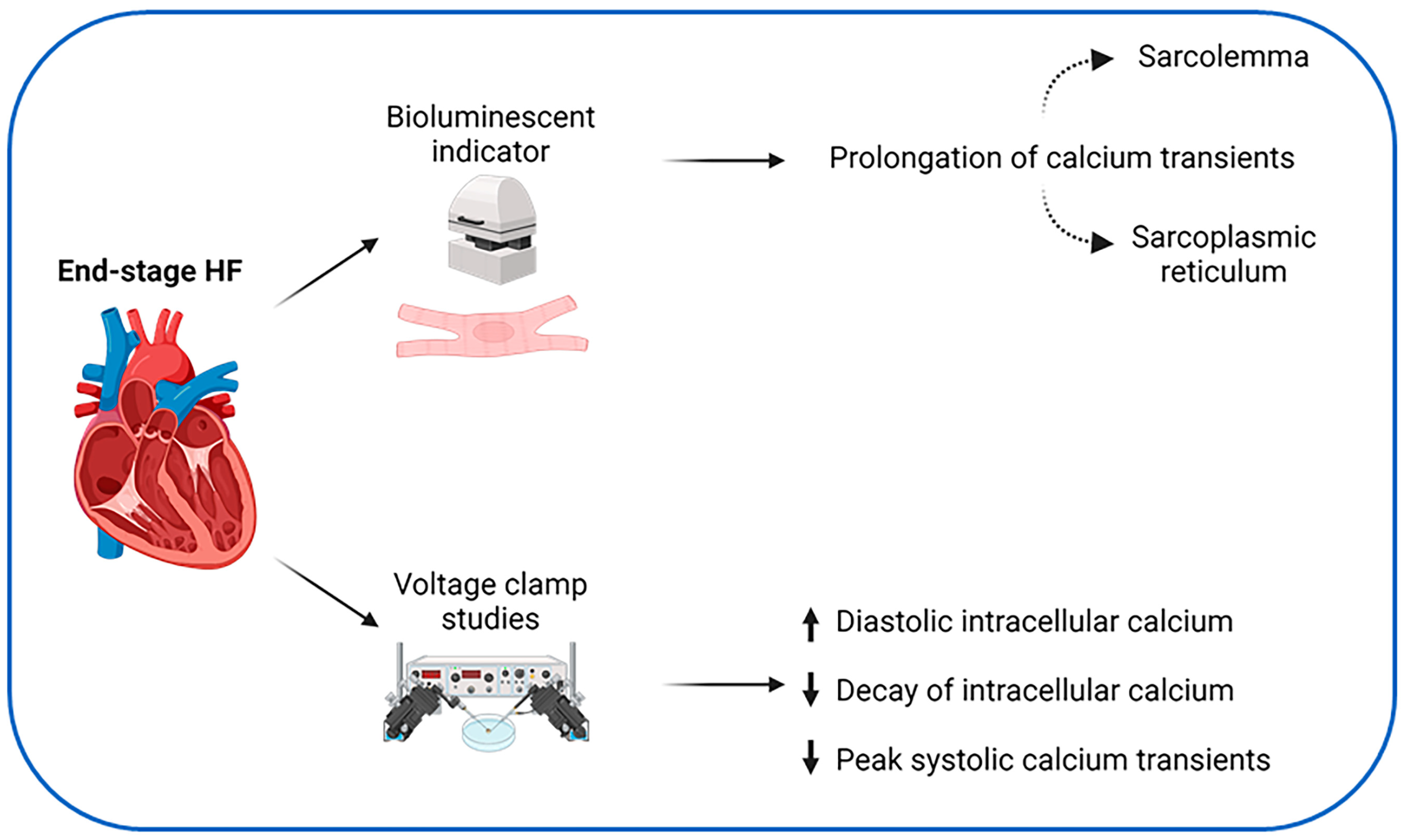
3. Calcium Handling in Heart Failure
4. Calcium Handling in Diabetic Cardiomyopathy
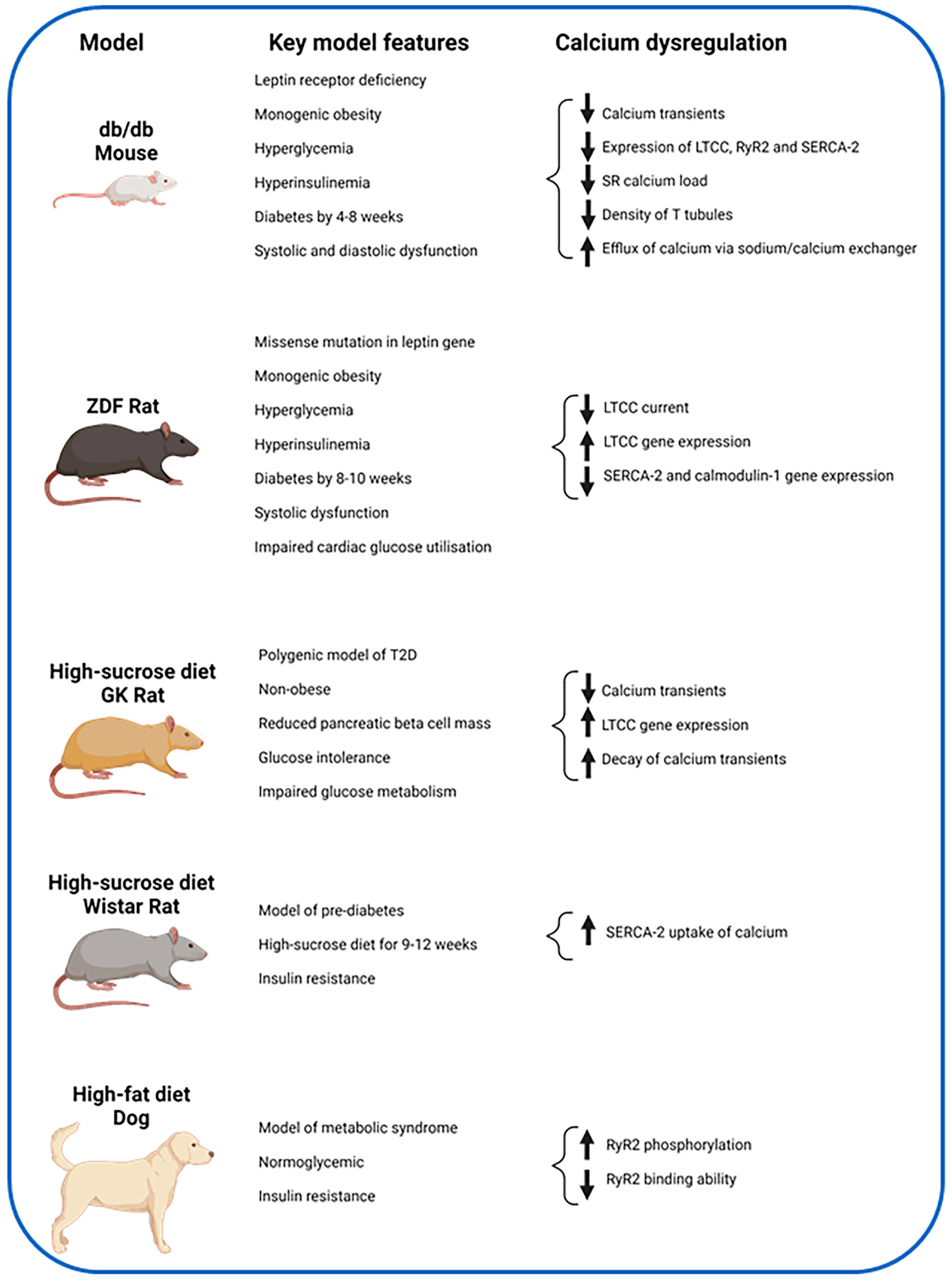
4.1. Animal Studies
4.1.1. LTCC
4.1.2. RyR2
4.1.3. SERCA-2
4.2. Human Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Model | Method | Key Findings |
|---|---|---|---|
| Ashrafi et al., 2017 [59] | Human T2D vs. non-TDM | T2D (n = 7) and non-T2D (n = 9) with severe aortic stenosis patients undergoing valve replacement. LV apical biopsy taken, quantitative PCR for genetic analysis. | Increase expression of gene encoding sodium/calcium |
| Drawnel et al., 2018 [64] | iPSC-CMs in diabetic environment vs. normal environment | Produced iPSC-CMs and placed in diabetic environment | Reduced calcium transient frequency and amplitude |
| Tang et al., 2020 [65] | iPSCs from T2D vs. HV 2 patients (T2D) vs. 5 controls (HV) | T2D (n = 2) and HV (n = 5). iPSCs generated from urine or skin cells | Reduced calcium transient amplitude, shorter transient duration, shorter decay, slower maximal rising rate, slower maximal decay rate. |
4.3. Mechanisms for Calcium Dysregulation in T2D
5. Potential for Magnetic Resonance Imaging to Study In Vivo Myocardial Calcium Handling
5.1. Cardiovascular Magnetic Resonance
5.2. Manganese and Cellular Function
5.3. Manganese-Enhanced MRI (MEMRI)
5.4. Clinical Studies Using MEMRI
5.5. MEMRI to Study Myocardial Infarction
5.6. MEMRI in Cardiomyopathies
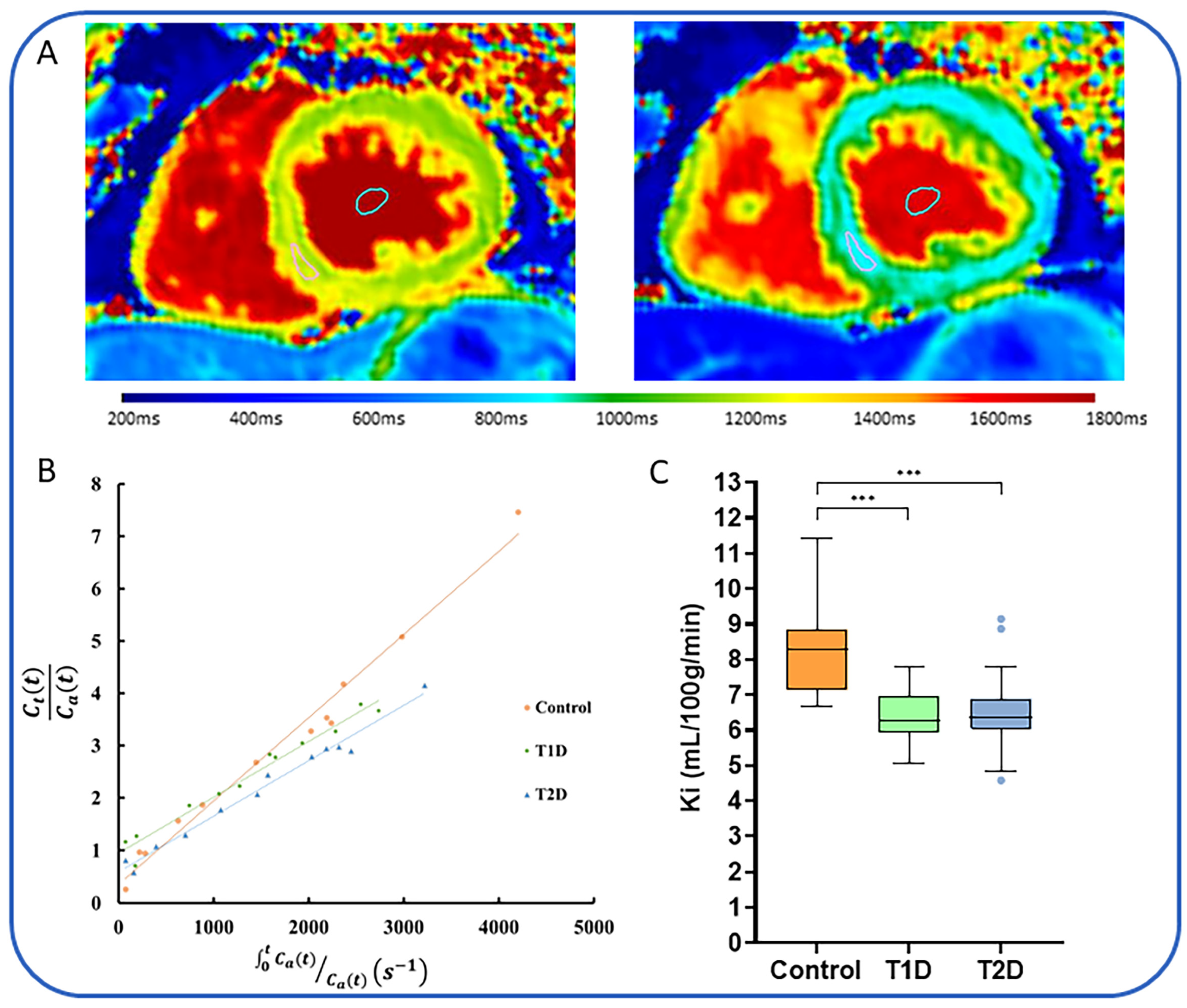
5.7. MEMRI in Diabetes
6. Calcium Handling as a Therapeutic Target in Diabetes
6.1. Glucagon-like Peptide-1 (GLP-1) Receptor Agonists
6.2. SGLT-2 Inhibitors
6.3. Calcium Sensitizers
6.4. Protein Kinases
6.5. Other Drug Therapy
6.6. Lifestyle Modification
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Koudstaal, S.; Pujades-Rodriguez, M.; Denaxas, S.; Gho, J.; Shah, A.D.; Yu, N.; Patel, R.S.; Gale, C.P.; Hoes, A.W.; Cleland, J.G.; et al. Prognostic burden of heart failure recorded in primary care, acute hospital admissions, or both: A population-based linked electronic health record cohort study in 2.1 million people. Eur. J. Heart Fail. 2017, 19, 1119–1127. [Google Scholar] [CrossRef]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Rawshani, A.; Rawshani, A.; Franzén, S.; Sattar, N.; Eliasson, B.; Svensson, A.-M.; Zethelius, B.; Miftaraj, M.; McGuire, D.K.; Rosengren, A.; et al. Risk Factors, Mortality, and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2018, 379, 633–644. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Lundbæk, K. Diabetic angiopathy: A specific vascular disease. Lancet 1954, 263, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.W.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association Of Glycaemia With Macrovascular And Microvascular Complications of Type 2 Diabetes (UKPDS 35): Prospective Observational Study. BMJ Br. Med. J. 2000, 321, 405–412. [Google Scholar] [CrossRef]
- Gottdiener, J.S.; Arnold, A.M.; Aurigemma, G.P.; Polak, J.F.; Tracy, R.P.; Kitzman, D.W.; Gardin, J.M.; Rutledge, J.E.; Boineau, R.C. Predictors of congestive heart failure in the elderly: The cardiovascular health study. J. Am. Coll. Cardiol. 2000, 35, 1628–1637. [Google Scholar] [CrossRef]
- Bertoni, A.G.; Tsai, A.; Kasper, E.K.; Brancati, F.L. Diabetes and idiopathic cardiomyopathy: A nationwide case-control study. Diabetes Care 2003, 26, 2791–2795. [Google Scholar] [CrossRef]
- Aronow, W.S.; Ahn, C. Incidence of heart failure in 2,737 older persons with and without diabetes mellitus. Chest 1999, 115, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Skali, H.; Shah, A.; Gupta, D.K.; Cheng, S.; Claggett, B.; Liu, J.; Bello, N.; Aguilar, D.; Vardeny, O.; Matsushita, K.; et al. Cardiac structure and function across the glycemic spectrum in elderly men and women free of prevalent heart disease: The Atherosclerosis Risk In the Community study. Circ. Heart Fail. 2015, 8, 448–454. [Google Scholar] [CrossRef] [PubMed]
- De Marco, M.; De Simone, G.; Roman, M.J.; Chinali, M.; Lee, E.T.; Calhoun, D.; Howard, B.V.; Devereux, R.B. Cardiac Geometry and Function in Diabetic or Prediabetic Adolescents and Young Adults: The Strong Heart Study. Diabetes Care 2011, 34, 2300–2305. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Larghat, A.M.; Swoboda, P.P.; Biglands, J.D.; Kearney, M.T.; Greenwood, J.P.; Plein, S. The microvascular effects of insulin resistance and diabetes on cardiac structure, function, and perfusion: A cardiovascular magnetic resonance study. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.N.; Wilmot, E.G.; Leggate, M.; Singh, A.; Yates, T.; Nimmo, M.; Khunti, K.; Horsfield, M.A.; Biglands, J.; Clarysse, P.; et al. Subclinical diastolic dysfunction in young adults with Type 2 diabetes mellitus: A multiparametric contrast-enhanced cardiovascular magnetic resonance pilot study assessing potential mechanisms. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Mahmod, M.; Piechnik, S.K.; Ariga, R.; Francis, J.M.; Rodgers, C.T.; Clarke, W.T.; Sabharwal, N.; Schneider, J.E.; Karamitsos, T.D.; et al. Relationship Between Left Ventricular Structural and Metabolic Remodeling in Type 2 Diabetes. Diabetes 2016, 65, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.K.; Thanigaraj, S.; Schechtman, K.B.; Pérez, J.E. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am. J. Cardiol. 2004, 93, 870–875. [Google Scholar] [CrossRef]
- Gulsin, G.S.; Swarbrick, D.J.; Athithan, L.; Brady, E.M.; Henson, J.; Baldry, E.; Argyridou, S.; Jaicim, N.B.; Squire, G.; Walters, Y.; et al. Effects of Low-Energy Diet or Exercise on Cardiovascular Function in Working-Age Adults with Type 2 Diabetes: A Prospective, Randomized, Open-Label, Blinded End Point Trial. Diabetes Care 2020, 43, 1300–1310. [Google Scholar] [CrossRef]
- Levelt, E.; Rodgers, C.T.; Clarke, W.T.; Mahmod, M.; Ariga, R.; Francis, J.M.; Liu, A.; Wijesurendra, R.S.; Dass, S.; Sabharwal, N.; et al. Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. Eur. Heart J. 2016, 37, 3461–3469. [Google Scholar] [CrossRef]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac Steatosis in Diabetes Mellitus. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef]
- Ringer, S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J. Physiol. 1883, 4, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A.; Fabiato, F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann. N. Y. Acad. Sci. 1978, 307, 491–522. [Google Scholar] [CrossRef] [PubMed]
- Bodi, I.; Mikala, G.; Koch, S.E.; Akhter, S.A.; Schwartz, A. The L-type calcium channel in the heart: The beat goes on. J. Clin. Investig. 2005, 115, 3306–3317. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.A.; Spinale, F.G. The structure and function of the cardiac myocyte: A review of fundamental concepts. J. Thorac. Cardiovasc. Surg. 1999, 118, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Barry, W.H.; Bridge, J.H. Intracellular calcium homeostasis in cardiac myocytes. Circulation 1993, 87, 1806–1815. [Google Scholar] [CrossRef]
- Toyofuku, T.; Curotto Kurzydlowski, K.; Narayanan, N.; MacLennan, D.H. Identification of Ser38 as the site in cardiac sarcoplasmic reticulum Ca(2+)-ATPase that is phosphorylated by Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 1994, 269, 26492–26496. [Google Scholar] [CrossRef]
- Ottolia, M.; Torres, N.; Bridge, J.H.; Philipson, K.D.; Goldhaber, J.I. Na/Ca exchange and contraction of the heart. J. Mol. Cell Cardiol. 2013, 61, 28–33. [Google Scholar] [CrossRef]
- Beard, N.A.; Laver, D.R.; Dulhunty, A.F. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef]
- Al Kury, L.T. Calcium Homeostasis in Ventricular Myocytes of Diabetic Cardiomyopathy. J. Diabetes Res. 2020, 2020, 1942086. [Google Scholar] [CrossRef]
- Halabi, A.; Yang, H.; Wright, L.; Potter, E.; Huynh, Q.; Negishi, K.; Marwick, T.H. Evolution of Myocardial Dysfunction in Asymptomatic Patients at Risk of Heart Failure. JACC Cardiovasc. Imaging 2021, 14, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Copelas, L.; MacKinnon, R.; Schoen, F.J.; Feldman, M.D.; Grossman, W.; Morgan, J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987, 61, 70–76. [Google Scholar] [CrossRef]
- Rasmussen, R.P.; Minobe, W.; Bristow, M.R. Calcium antagonist binding sites in failing and nonfailing human ventricular myocardium. Biochem. Pharmacol. 1990, 39, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Näbauer, M.; Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Piacentino, V.; Weber, C.R.; Chen, X.; Weisser-Thomas, J.; Margulies, K.B.; Bers, D.M.; Houser, S.R. Cellular Basis of Abnormal Calcium Transients of Failing Human Ventricular Myocytes. Circ. Res. 2003, 92, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Schröder, F.; Handrock, R.; Beuckelmann, D.J.; Hirt, S.; Hullin, R.; Priebe, L.; Schwinger, R.H.; Weil, J.; Herzig, S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998, 98, 969–976. [Google Scholar] [CrossRef]
- Goonasekera, S.A.; Hammer, K.; Auger-Messier, M.; Bodi, I.; Chen, X.; Zhang, H.; Reiken, S.; Elrod, J.W.; Correll, R.N.; York, A.J.; et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 2012, 122, 280–290. [Google Scholar] [CrossRef]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef]
- Kiss, E.; Ball, N.A.; Kranias, E.G.; Walsh, R.A. Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)-ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ. Res. 1995, 77, 759–764. [Google Scholar] [CrossRef]
- Kuo, T.H.; Zhu, L.; Golden, K.; Marsh, J.D.; Bhattacharya, S.K.; Liu, B.-F. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol. Cell. Biochem. 2002, 238, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Le, C.; Shen, X.; Røe, Å.T.; Hou, Y.; Manfra, O.; Silva, G.J.J.; van Hout, I.; Norden, E.S.; Aronsen, J.M.; et al. Etiology-Dependent Impairment of Diastolic Cardiomyocyte Calcium Homeostasis in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Enhancing Mitochondrial Ca2+ Uptake in Myocytes From Failing Hearts Restores Energy Supply and Demand Matching. Circ. Res. 2008, 103, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Huo, Y. Changes of calcium cycling in HFrEF and HFpEF. Mechanobiol. Med. 2023, 1, 100001. [Google Scholar] [CrossRef]
- McHugh, K.; DeVore, A.D.; Wu, J.; Matsouaka, R.A.; Fonarow, G.C.; Heidenreich, P.A.; Yancy, C.W.; Green, J.B.; Altman, N.; Hernandez, A.F. Heart Failure With Preserved Ejection Fraction and Diabetes. J. Am. Coll. Cardiol. 2019, 73, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Semeniuk, L.M.; Kryski, A.J.; Severson, D.L. Echocardiographic assessment of cardiac function in diabeticdb/db and transgenic db/db-hGLUT4 mice. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H976–H982. [Google Scholar] [CrossRef]
- van den Brom, C.E.; Bosmans, J.W.; Vlasblom, R.; Handoko, L.M.; Huisman, M.C.; Lubberink, M.; Molthoff, C.F.; Lammertsma, A.A.; Ouwens, M.D.; Diamant, M.; et al. Diabetic cardiomyopathy in Zucker diabetic fatty rats: The forgotten right ventricle. Cardiovasc. Diabetol. 2010, 9, 25. [Google Scholar] [CrossRef]
- King, A.J.F. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [CrossRef]
- Wold, L.E.; Dutta, K.; Mason, M.M.; Ren, J.; Cala, S.E.; Schwanke, M.L.; Davidoff, A.J. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell Cardiol. 2005, 39, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Dincer, U.D.; Araiza, A.; Knudson, J.D.; Shao, C.H.; Bidasee, K.R.; Tune, J.D. Dysfunction of cardiac ryanodine receptors in the metabolic syndrome. J. Mol. Cell Cardiol. 2006, 41, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gómez, A.M. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Stølen, T.O.; Høydal, M.A.; Kemi, O.J.; Catalucci, D.; Ceci, M.; Aasum, E.; Larsen, T.; Rolim, N.; Condorelli, G.; Smith, G.L.; et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 2009, 105, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Howarth, F.C.; Qureshi, M.A.; Hassan, Z.; Al Kury, L.T.; Isaev, D.; Parekh, K.; Yammahi, S.R.; Oz, M.; Adrian, T.E.; Adeghate, E. Changing pattern of gene expression is associated with ventricular myocyte dysfunction and altered mechanisms of Ca2+ signalling in young type 2 Zucker diabetic fatty rat heart. Exp. Physiol. 2011, 96, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Salem, K.A.; Adrian, T.E.; Qureshi, M.A.; Parekh, K.; Oz, M.; Howarth, F.C. Shortening and intracellular Ca2+ in ventricular myocytes and expression of genes encoding cardiac muscle proteins in early onset type 2 diabetic Goto-Kakizaki rats. Exp. Physiol. 2012, 97, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Gaber, E.M.; Jayaprakash, P.; Qureshi, M.A.; Parekh, K.; Oz, M.; Adrian, T.E.; Howarth, F.C. Effects of a sucrose-enriched diet on the pattern of gene expression, contraction and Ca(2+) transport in Goto-Kakizaki type 2 diabetic rat heart. Exp. Physiol. 2014, 99, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T.; Baughman, K.L.; Feldman, A.M.; Frustaci, A.; Jessup, M.; Kuhl, U.; Levine, G.N.; Narula, J.; Starling, R.C.; Towbin, J.; et al. The Role of Endomyocardial Biopsy in the Management of Cardiovascular Disease. Circulation 2007, 116, 2216–2233. [Google Scholar] [CrossRef]
- Ashrafi, R.; Modi, P.; Oo, A.Y.; Pullan, D.M.; Jian, K.; Zhang, H.; Gerges, J.Y.; Hart, G.; Boyett, M.R.; Davis, G.K.; et al. Arrhythmogenic gene remodelling in elderly patients with type 2 diabetes with aortic stenosis and normal left ventricular ejection fraction. Exp. Physiol. 2017, 102, 1424–1434. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Fraser, S.T.; Nowak, N.; Semsarian, C. Generation of induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient with the pathogenic variant p.Val698Ala in beta-myosin heavy chain (MYH7) gene. Stem Cell Res. 2017, 20, 88–90. [Google Scholar] [CrossRef]
- Granéli, C.; Hicks, R.; Brolén, G.; Synnergren, J.; Sartipy, P. Diabetic Cardiomyopathy Modelling Using Induced Pluripotent Stem Cell Derived Cardiomyocytes: Recent Advances and Emerging Models. Stem Cell Rev. Rep. 2019, 15, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gérard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014, 9, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, H.; Dai, B.; Wang, X.; Zhou, D.; Shen, J.; Guo, F.; Wang, J.; Zhou, J.; Wang, H.; et al. Human induced pluripotent stem cell-derived cardiomyocytes reveal abnormal TGFb; signaling in type 2 diabetes mellitus. J. Mol. Cell. Cardiol. 2020, 142, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Cameron, F.; Tiwari, S.; Mannering, S.I.; Elefanty, A.G.; Stanley, E.G. Modeling Type 1 Diabetes Using Pluripotent Stem Cell Technology. Front. Endocrinol. 2021, 12, 635662. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Hegab, Z.; Mohamed, T.M.A.; Stafford, N.; Mamas, M.; Cartwright, E.J.; Oceandy, D. Advanced glycation end products reduce the calcium transient in cardiomyocytes by increasing production of reactive oxygen species and nitric oxide. FEBS Open Bio 2017, 7, 1672–1685. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Nallani, K.; Yu, Y.; Cocklin, R.R.; Zhang, Y.; Wang, M.; Dincer, U.D.; Besch, H.R., Jr. Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes 2003, 52, 1825–1836. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- Ripley, D.P.; Musa, T.A.; Dobson, L.E.; Plein, S.; Greenwood, J.P. Cardiovascular magnetic resonance imaging: What the general cardiologist should know. Heart 2016, 102, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.P. Cardiovascular magnetic resonance physics for clinicians: Part I. J. Cardiovasc. Magn. Reson. 2010, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Laniado, M.; Weinmann, H.J.; Schörner, W.; Felix, R.; Speck, U. First use of GdDTPA/dimeglumine in man. Physiol. Chem. Phys. Med. NMR 1984, 16, 157–165. [Google Scholar] [PubMed]
- Strich, G.; Hagan, P.L.; Gerber, K.H.; Slutsky, R.A. Tissue distribution and magnetic resonance spin lattice relaxation effects of gadolinium-DTPA. Radiology 1985, 154, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Brasch, R.C. Work in progress: Methods of contrast enhancement for NMR imaging and potential applications. A subject review. Radiology 1983, 147, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Currie, G.M. Pharmacology, Part 5: CT and MRI Contrast Media. J. Nucl. Med. Technol. 2019, 47, 189–202. [Google Scholar] [CrossRef]
- Ochi, R. The slow inward current and the action of manganese ions in guinea-pig’s myocardium. Pflug. Arch. 1970, 316, 81–94. [Google Scholar] [CrossRef]
- Ochi, R. Manganese-dependent propagated action potentials and their depression by electrical stimulation in guinea-pig myocardium perfused by sodium-free media. J. Physiol. 1976, 263, 139–156. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A.; Berkoff, H.A. Cellular manganese uptake by the isolated perfused rat heart: A probe for the sarcolemma calcium channel. J. Mol. Cell. Cardiol. 1981, 13, 823–832. [Google Scholar] [CrossRef]
- Wendland, M.F. Applications of manganese-enhanced magnetic resonance imaging (MEMRI) to imaging of the heart. NMR Biomed. 2004, 17, 581–594. [Google Scholar] [CrossRef]
- Mendonça-Dias, M.H.; Gaggelli, E.; Lauterbur, P.C. Paramagnetic contrast agents in nuclear magnetic resonance medical imaging. Semin. Nucl. Med. 1983, 13, 364–376. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public Statement on Teslascan: Withdrawal of the Marketing Authorisation in the European Union. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/teslascan (accessed on 21 August 2021).
- Wolf, G.L.; Baum, L. Cardiovascular toxicity and tissue proton T1 response to manganese injection in the dog and rabbit. AJR Am. J. Roentgenol. 1983, 141, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Takeya, K.; Reiter, M. Effect of divalent manganese ions on action potential and contractility of cardiac muscle. Naunyn Schmiedebergs Arch. Pharmacol. 1972, 275, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Brurok, H.; Schjøtt, J.; Berg, K.; Karlsson, J.O.; Jynge, P. Manganese and the heart: Acute cardiodepression and myocardial accumulation of manganese. Acta Physiol. Scand. 1997, 159, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.C.; Pautler, R.G.; MacGowan, G.A.; Koretsky, A.P. Manganese-enhanced MRI of mouse heart during changes in inotropy. Magn. Reson. Med. 2001, 46, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Skjold, A.; Vangberg, T.R.; Kristoffersen, A.; Haraldseth, O.; Jynge, P.; Larsson, H.B. Relaxation enhancing properties of MnDPDP in human myocardium. J. Magn. Reson. Imaging 2004, 20, 948–952. [Google Scholar] [CrossRef]
- Skjold, A.; Kristoffersen, A.; Vangberg, T.R.; Haraldseth, O.; Jynge, P.; Larsson, H.B. An apparent unidirectional influx constant for manganese as a measure of myocardial calcium channel activity. J. Magn. Reson. Imaging 2006, 24, 1047–1055. [Google Scholar] [CrossRef]
- Skjold, A.; Amundsen, B.H.; Wiseth, R.; Støylen, A.; Haraldseth, O.; Larsson, H.B.; Jynge, P. Manganese dipyridoxyl-diphosphate (MnDPDP) as a viability marker in patients with myocardial infarction. J. Magn. Reson. Imaging 2007, 26, 720–727. [Google Scholar] [CrossRef]
- Spath, N.B.; Singh, T.; Papanastasiou, G.; Kershaw, L.; Baker, A.H.; Janiczek, R.L.; Gulsin, G.S.; Dweck, M.R.; McCann, G.; Newby, D.E.; et al. Manganese-enhanced magnetic resonance imaging in dilated cardiomyopathy and hypertrophic cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 1463–1472. [Google Scholar] [CrossRef]
- Spath, N.B.; Singh, T.; Papanastasiou, G.; Baker, A.; Janiczek, R.J.; McCann, G.P.; Dweck, M.R.; Kershaw, L.; Newby, D.E.; Semple, S. Assessment of stunned and viable myocardium using manganese-enhanced MRI. Open Heart 2021, 8, e001646. [Google Scholar] [CrossRef]
- Singh, T.; Joshi, S.; Kershaw, L.E.; Baker, A.H.; McCann, G.P.; Dawson, D.K.; Dweck, M.R.; Semple, S.I.; Newby, D.E. Manganese-Enhanced Magnetic Resonance Imaging in Takotsubo Syndrome. Circulation 2022, 146, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Dattani, A.; Joshi, S.; Yeo, J.L.; Singh, A.; Brady, E.M.; Parke, K.S.; Arnold, J.R.; Singh, T.; Kershaw, L.E.; Spath, N.B.; et al. Impaired Myocardial Calcium Uptake in Patients with Diabetes Mellitus: A Manganese-Enhanced Cardiac Magnetic Resonance Study. JACC Cardiovasc. Imaging 2023, 16, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792. [Google Scholar] [CrossRef] [PubMed]
- Partnership, H.Q.I. National diabetes Audit 2015–2016. Available online: http://content.digital.nhs.uk/nda (accessed on 13 October 2023).
- Piepoli, M.F.; Adamo, M.; Barison, A.; Bestetti, R.B.; Biegus, J.; Bohm, M.; Butler, J.; Carapetis, J.; Ceconi, C.; Chioncel, O.; et al. Preventing heart failure: A position paper of the Heart Failure Association in collaboration with the European Association of Preventive Cardiology. Eur. J. Heart Fail. 2022, 24, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-H.; Chen, Y.-C.; Lee, T.-I.; Kao, Y.-H.; Chazo, T.-F.; Chen, S.-A.; Chen, Y.-J. Glucagon-like peptide-1 regulates calcium homeostasis and electrophysiological activities of HL-1 cardiomyocytes. Peptides 2016, 78, 91–98. [Google Scholar] [CrossRef]
- Xiao, Y.F.; Nikolskaya, A.; Jaye, D.A.; Sigg, D.C. Glucagon-like peptide-1 enhances cardiac L-type Ca2+ currents via activation of the cAMP-dependent protein kinase A pathway. Cardiovasc. Diabetol. 2011, 10, 6. [Google Scholar] [CrossRef]
- Khan, M.S.; Fonarow, G.C.; McGuire, D.K.; Hernandez, A.F.; Vaduganathan, M.; Rosenstock, J.; Handelsman, Y.; Verma, S.; Anker, S.D.; McMurray, J.J.V.; et al. Glucagon-Like Peptide 1 Receptor Agonists and Heart Failure. Circulation 2020, 142, 1205–1218. [Google Scholar] [CrossRef]
- Kosiborod, M.N.; Abildstrøm, S.Z.; Borlaug, B.A.; Butler, J.; Christensen, L.; Davies, M.; Hovingh, K.G.; Kitzman, D.W.; Lindegaard, M.L.; Møller, D.V.; et al. Design and Baseline Characteristics of STEP-HFpEF Program Evaluating Semaglutide in Patients With Obesity HFpEF Phenotype. JACC Heart Fail. 2023, 11, 1000–1010. [Google Scholar] [CrossRef]
- Marfella, R.; Scisciola, L.; D’Onofrio, N.; Maiello, C.; Trotta, M.C.; Sardu, C.; Panarese, I.; Ferraraccio, F.; Capuano, A.; Barbieri, M.; et al. Sodium-glucose cotransporter-2 (SGLT2) expression in diabetic and non-diabetic failing human cardiomyocytes. Pharmacol. Res. 2022, 184, 106448. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Kass, D.A.; Solaro, R.J. Mechanisms and Use of Calcium-Sensitizing Agents in the Failing Heart. Circulation 2006, 113, 305–315. [Google Scholar] [CrossRef]
- Akhtar, M.S.; Pillai, K.K.; Hassan, M.Q.; Dhyani, N.; Ismail, M.V.; Najmi, A.K. Levosimendan reduces myocardial damage and improves cardiodynamics in streptozotocin induced diabetic cardiomyopathy via SERCA2a/NCX1 pathway. Life Sci. 2016, 153, 55–65. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Bowling, N.; Walsh, R.A.; Song, G.; Estridge, T.; Sandusky, G.E.; Fouts, R.L.; Mintze, K.; Pickard, T.; Roden, R.; Bristow, M.R.; et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 1999, 99, 384–391. [Google Scholar] [CrossRef]
- Takeishi, Y.; Chu, G.; Kirkpatrick, D.M.; Li, Z.; Wakasaki, H.; Kranias, E.G.; King, G.L.; Walsh, R.A. In vivo phosphorylation of cardiac troponin I by protein kinase Cbeta2 decreases cardiomyocyte calcium responsiveness and contractility in transgenic mouse hearts. J. Clin. Investig. 1998, 102, 72–78. [Google Scholar] [CrossRef]
- Banerjee, S.; Khan, M.K.A. Chapter 23—Calcium calmodulin-dependent protein kinase as a potential drug target. In Protein Kinase Inhibitors; Hassan, M.I., Noor, S., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 657–670. [Google Scholar]
- Aditya, S.; Rattan, A. Istaroxime: A rising star in acute heart failure. J. Pharmacol. Pharmacother. 2012, 3, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Torre, E.; Arici, M.; Lodrini, A.M.; Ferrandi, M.; Barassi, P.; Hsu, S.C.; Chang, G.J.; Boz, E.; Sala, E.; Vagni, S.; et al. SERCA2a stimulation by istaroxime improves intracellular Ca2+ handling and diastolic dysfunction in a model of diabetic cardiomyopathy. Cardiovasc. Res. 2022, 118, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ren, L.; Liu, X.; Sun, X.; Dong, C.; Jiang, Y.; Qin, Y.; Qu, H.; Jiao, J.; Wang, S.; et al. Ranolazine protects against diabetic cardiomyopathy by activating the NOTCH1/NRG1 pathway. Life Sci. 2020, 261, 118306. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, K.; Boulé, N.; Henson, J.; Chevalier, S.; Redman, E.; Chan, D.; McCarthy, M.; Champagne, J.; Arsenyadis, F.; Rees, J.; et al. Remission of type 2 diabetes and improved diastolic function by combining structured exercise with meal replacement and food reintroduction among young adults: The RESET for REMISSION randomised controlled trial protocol. BMJ Open 2022, 12, e063888. [Google Scholar] [CrossRef]




| Author, Year | Model | Method | Key Findings |
|---|---|---|---|
| Wold et al., 2005 [51] | High sucrose diet rats (insulin resistant model) vs. controls | 9–12 weeks dietary intervention of high sucrose diet. Fluo-3, immunoblot assays | Insulin resistant rats have slower cytosolic calcium removal and slower SERCA2 calcium uptake. |
| Dincer et al., 2006 [52] | High-fat diet (metabolic syndrome model) vs. normal diet in dogs (control). | Six-week dietary intervention. RNA samples from RA, RV and LV, reverse transcription and RT-PCR. Western blot analyses. Ser2809 phospho-specific antibodies and [3H]ryanodine for RyR2 assessment. | Increased RyR2 phosphorylation at Ser2809 and reduced ability of the RyR2 bind to [3H]ryanodine. No change in the gene expression of RyR2. |
| Pereira et al., 2006 [53] | Diabetic mouse (db/db) vs. control mouse (+/+). | Two-photon microscopy, confocal microscopy, Fluo-3, whole-cell patch clamp technique, western blots | Decreased intracellular calcium transients, LTCC expression, calcium current, SR calcium load, ryanodine receptors, and increased calcium efflux via sodium/calcium exchanger |
| Stølen et al., 2009 [54] | Diabetic mouse (db/db) vs. heterozygote controls | Fura-2 and Fluo-3 indicators, bipolar electric pulses, western blots, RT-PCR, exercise training, echocardiography | Sedentary db/db mice had lower calcium release, lower SR calcium load, slower calcium decay, reduced T tubule density, reduced expression of SERCA-2a. All features improved with exercise training. |
| Howarth et al., 2011 [55] | ZDF rat vs. age-matched control lean | Fura-2, whole cell patch clamp techniques, RNA sampling from LV apex with reverse transcription and gene expression assays. | Upregulation of genes for LTCC subunits. Downregulation of genes for SERCA2 and Calmodulin 1. Reduced LTCC current in ZDF rats, prolonged inactivation of LTCC current and prolonged time to peak calcium transient. No change in SR release, uptake and calcium content. |
| Salem et al., 2012 [56] | GK rats vs. Wistar control rats | Fura-2, RNA sampling from LV apex with reverse transcription and gene expression assays | No change in LTCC subunit gene expression. Upregulation of TTCC, potassium and sodium channel gene expression. No change in intracellular calcium amplitude but faster decay of calcium transient seen. |
| Gaber et al., 2014 [57] | GK vs. GK-sucrose vs. Control (Wistar) vs. Control-sucrose rats | Eight-month intervention of sucrose-enriched water. Fura-2 used. Ventricular RNA samples with reverse transcription and gene expression assays | Prolonged time to peak shortening of myocyte in GK group, reduced amplitude of myocyte shortening and reduced amplitude of calcium transients in the sucrose groups. Upregulation of genes for LTCC subunits in GK rats and RyR2 in control-sucrose rats. |
| Author, Year | Participants | Inclusion Criteria | Exclusion Criteria | Method | Key Findings |
|---|---|---|---|---|---|
| Skjold et al., 2007 [89] | 10 | Males (n = 8) and females, age 37–75, MI within 12 weeks with PCI. | Given 5 µmol/kg of MnDPDP over 5 min, 1.5T scanner used to obtain images pre- and post-contrast. | Higher T1 relaxation rates in non-infarcted myocardium vs. infarcted myocardium. | |
| Spath et al., 2020 [90] | 47 | HVs (n = 20), non-ischaemic DCM (n = 10) and HCM (n = 17) | NYHA class IV, contraindications to MRI or MnDPDP. | Open-label, observational cohort study. Gadolinium-enhanced MRI and MEMRI >48 h apart. T1 mapping performed using 3T scanner every 2.5 min. MnDPDP administered at dose of 5 µmol/kg at rate of 1 mL/min. | Patients had lower mean reductions in T1 values and lower Ki following MnDPDP compared to HVs. |
| Spath et al., 2021 [91] | 40 | HV (n = 20). STEMI patients (n = 20) with proven LMS, LAD or multi-vessel disease, clinically stable, EF < 50%, | NYHA class IV, contraindications to MRI or MnDPDP. | Open-label, observational cohort study. Gadolinium-enhanced MRI and MEMRI >48 h apart, within 7 days of revascularisation and at 3 months. T1 mapping performed using 3T scanner every 2.5 min. MnDPDP administered at dose of 5 µmol/kg at rate of 1 mL/min. | Infarcted myocardium had higher T1 values at 40 min compared to remote myocardium and HVs. Ki values were lower in infarcted compared to peri-infarct myocardium, which were also lower than remote myocardium. |
| Singh et al., 2022 [92] | 40 | HV (n = 20) and Takotsubo cardiomyopathy patients (n = 20) | Prospective, case-control study using MEMRI. T1 mapping performed using 3T scanner every 2.5 min. MnDPDP administered at dose of 5 µmol/kg at rate of 1 mL/min. | Patients with Takotsubo had lower mean Ki which persisted despite recovery of LV systolic function. | |
| Dattani et al., 2023 [93] | 60 | HV (n = 11), T1D (n = 19), T2D (n = 30). | Symptoms or history of cardiac disease | Prospective, case-control study using MEMRI. T1 mapping performed using 3T scanner every 2.5 min. MnDPDP administered at dose of 5 µmol/kg at rate of 1 mL/min. | Patients with T1D and T2D had lower mean Ki compared to controls. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dattani, A.; Singh, A.; McCann, G.P.; Gulsin, G.S. Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target. J. Cardiovasc. Dev. Dis. 2024, 11, 12. https://doi.org/10.3390/jcdd11010012
Dattani A, Singh A, McCann GP, Gulsin GS. Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target. Journal of Cardiovascular Development and Disease. 2024; 11(1):12. https://doi.org/10.3390/jcdd11010012
Chicago/Turabian StyleDattani, Abhishek, Anvesha Singh, Gerry P. McCann, and Gaurav S. Gulsin. 2024. "Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target" Journal of Cardiovascular Development and Disease 11, no. 1: 12. https://doi.org/10.3390/jcdd11010012
APA StyleDattani, A., Singh, A., McCann, G. P., & Gulsin, G. S. (2024). Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target. Journal of Cardiovascular Development and Disease, 11(1), 12. https://doi.org/10.3390/jcdd11010012