
Cardiac and Renal Fibrosis, the Silent Killer in the Cardiovascular Continuum: An Up-to-Date

 , , , , , ,
, , , , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms and Molecules Involved in Activation and Promotion of Fibrosis
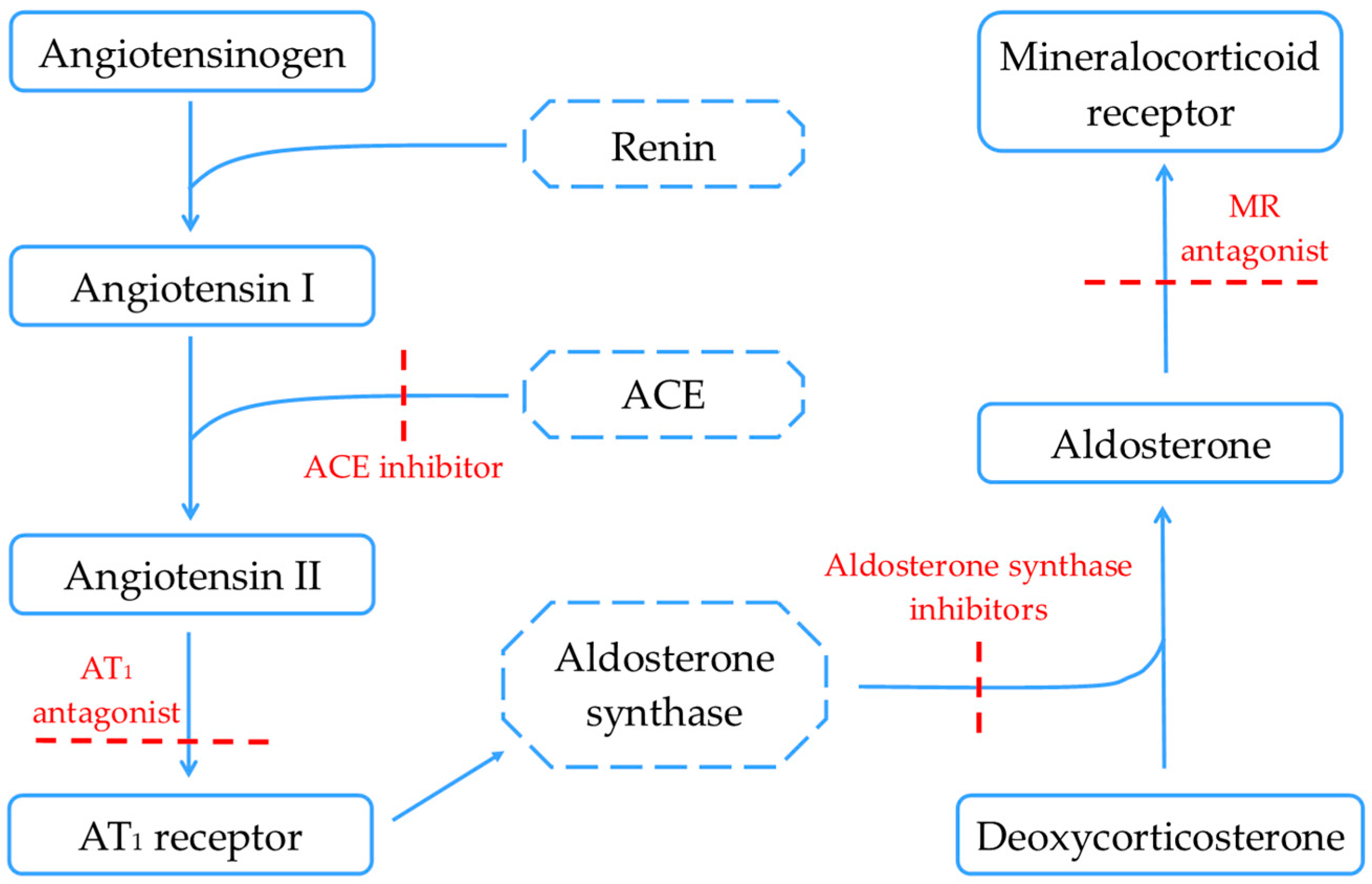
2.1. Renin–Angiotensin–Aldosterone System
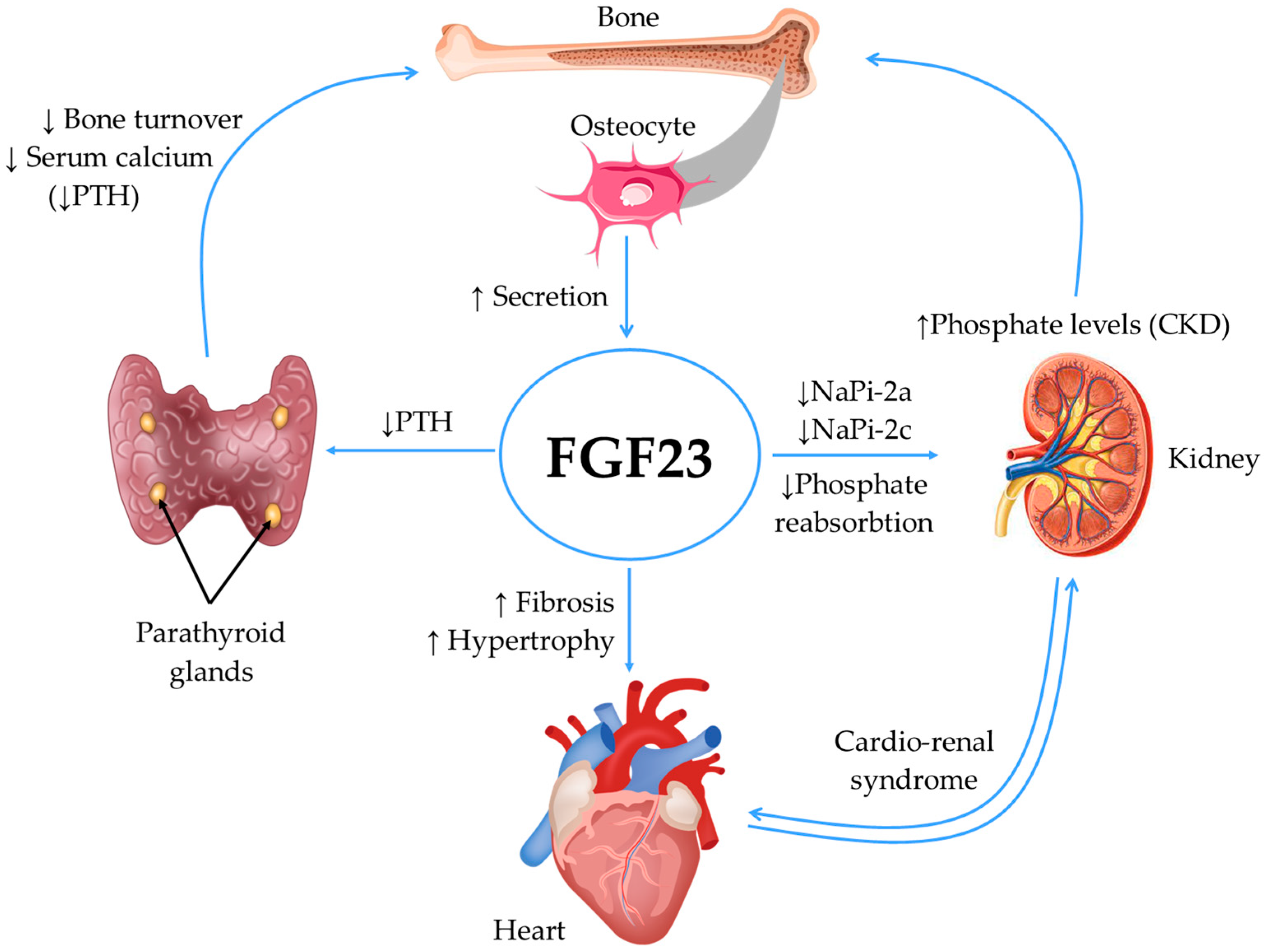
2.2. Fibroblast Growth Factor 23 (FGF23) and Klotho Protein
2.3. Kidney Injury Molecule-1 (KIM-1)
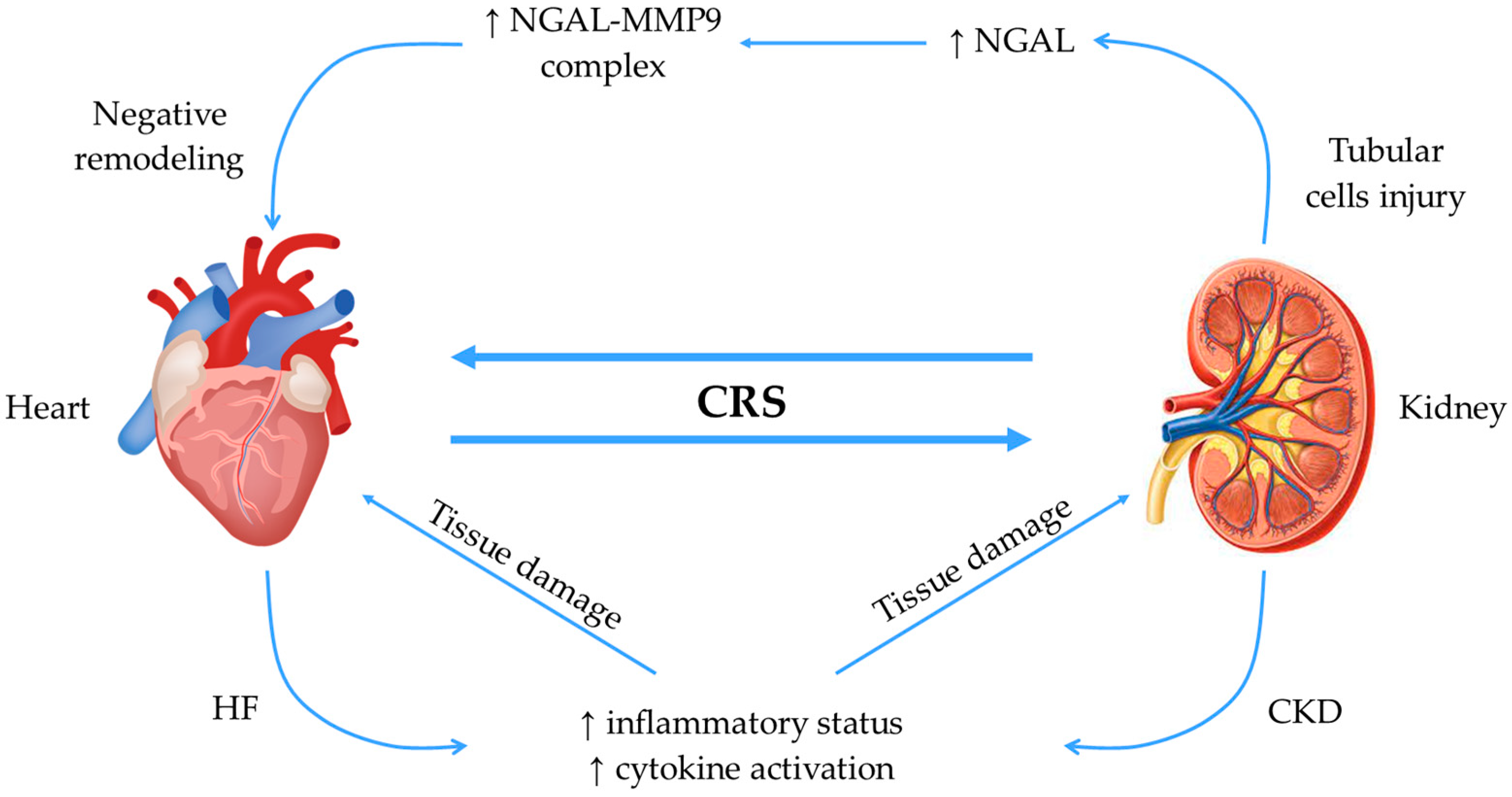
2.4. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
2.5. Relaxin
2.6. Collagen Biomarkers
2.7. Other Mechanisms and Pathways
2.7.1. Redox Signaling and Oxidative Stress
2.7.2. Inflammasomes
3. Emerging Therapeutic Targets
3.1. RAAS Inhibitors—Future Perspectives
3.2. Serelaxin
3.3. Neutralizing Interleukin-11 (IL-11) Antibodies
3.4. Individualized Therapeutic Approach
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Preeti, J.; Alexandre, M.; Pupalan, I.; CMerlin, T.; Claudio, R. Chronic Heart Failure and Comorbid Renal Dysfunction—A Focus on Type 2 Cardiorenal Syndrome. Curr. Cardiol. Rev. 2016, 12, 186–194. [Google Scholar] [CrossRef]
- Boriani, G.; Savelieva, I.; Dan, G.A.; Deharo, J.C.; Ferro, C.; Israel, C.W.; Lane, D.A.; La Manna, G.; Morton, J.; Mitjans, A.M.; et al. Chronic kidney disease in patients with cardiac rhythm disturbances or implantable electrical devices: Clinical significance and implications for decision making-a position paper of the European Heart Rhythm Association endorsed by the Heart Rhythm Society and the Asia Pacific Heart Rhythm Society. Ep Eur. 2015, 17, 1169–1196. [Google Scholar]
- Clementi, A.; Virzì, G.M.; Goh, C.Y.; Cruz, D.N.; Granata, A.; Vescovo, G.; Ronco, C. Cardiorenal syndrome type 4: A review. Cardiorenal Med. 2013, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.; Anker, S.D.; Mebazaa, A.; Palazzuoli, A.; Vescovo, G.; Bellomo, R.; Ponikowski, P.; Anand, I.; Aspromonte, N.; Bagshaw, S.; et al. ADQI 7: The clinical management of the Cardio-Renal syndromes: Work group statements from the 7th ADQI consensus conference. Nephrol. Dial. Transplant. 2010, 25, 2077–2089. [Google Scholar] [CrossRef]
- House, A.A.; Anand, I.; Bellomo, R.; Cruz, D.; Bobek, I.; Anker, S.D.; Aspromonte, N.; Bagshaw, S.; Berl, T.; Daliento, L.; et al. Definition and classification of Cardio-Renal Syndromes: Workgroup statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Hulshoff, M.S.; Rath, S.K.; Xu, X.; Zeisberg, M.; Zeisberg, E.M. Causal Connections From Chronic Kidney Disease to Cardiac Fibrosis. Semin. Nephrol. 2018, 38, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Romero-González, G.; González, A.; López, B.; Ravassa, S.; Díez, J. Heart failure in chronic kidney disease: The emerging role of myocardial fibrosis. Nephrol. Dial. Transplant. 2020, 37, 817–824. [Google Scholar] [CrossRef]
- Alshahrani, S. Renin-angiotensin-aldosterone pathway modulators in chronic kidney disease: A comparative review. Front. Pharmacol. 2023, 14, 1101068. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Schmidt-Ott, K.M.; Vescovo, G.; House, A.A.; Kellum, J.A.; Ronco, C.; McCullough, P.A.; Acute Dialysis Quality Initiative. Pathophysiology of Cardiorenal Syndrome Type 2 in Stable Chronic Heart Failure: Workgroup Statements from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). In ADQI Consensus on AKI Biomarkers and Cardiorenal Syndromes; Kellum, J.A., McCullough, P.A., Mehta, R.L., Murray, P.T., Ronco, C., Eds.; Karger Publishers: Basel, Switzerland, 2013; Volume 182. [Google Scholar]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- Huang, W.; Xu, C.; Kahng, K.W.; Noble, N.A.; Border, W.A.; Huang, Y. Aldosterone and TGF-beta1 synergistically increase PAI-1 and decrease matrix degradation in rat renal mesangial and fibroblast cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F1287–F1295. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Wacker, M.J. FGF23 production by osteocytes. Pediatr. Nephrol. 2013, 28, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Kishimoto, H.; Tokumoto, M. Direct and indirect effects of fibroblast growth factor 23 on the heart. Front. Endocrinol. 2023, 14, 1059179. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Li, X.; Li, Q.; Lin, H.; Chen, Z.; Xie, J.; Xuan, W.; Liao, W.; Bin, J.; Huang, X.; et al. FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 2016, 7, 64649–64664. [Google Scholar] [CrossRef]
- Saito, T.; Mizobuchi, M.; Kato, T.; Ogata, H.; Koiwa, F.; Honda, H. Fibroblast Growth Factor 23 Exacerbates Cardiac Fibrosis in Deoxycorticosterone Acetate-Salt Mice With Hypertension. Lab. Investig. 2023, 103, 100003. [Google Scholar] [CrossRef]
- Scialla, J.J.; Wolf, M. Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat. Rev. Nephrol. 2014, 10, 268–278. [Google Scholar] [CrossRef]
- McGovern, A.P.; de Lusignan, S.; van Vlymen, J.; Liyanage, H.; Tomson, C.R.; Gallagher, H.; Rafiq, M.; Jones, S. Serum phosphate as a risk factor for cardiovascular events in people with and without chronic kidney disease: A large community based cohort study. PLoS ONE 2013, 8, e74996. [Google Scholar] [CrossRef]
- Lee, T.W.; Chung, C.C.; Lee, T.I.; Lin, Y.K.; Kao, Y.H.; Chen, Y.J. Fibroblast Growth Factor 23 Stimulates Cardiac Fibroblast Activity through Phospholipase C-Mediated Calcium Signaling. Int. J. Mol. Sci. 2021, 23, 166. [Google Scholar] [CrossRef] [PubMed]
- Leifheit-Nestler, M.; Kirchhoff, F.; Nespor, J.; Richter, B.; Soetje, B.; Klintschar, M.; Heineke, J.; Haffner, D. Fibroblast growth factor 23 is induced by an activated renin-angiotensin-aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol. Dial. Transplant. 2018, 33, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Gabbin, B.; Meraviglia, V.; Mummery, C.L.; Rabelink, T.J.; van Meer, B.J.; van den Berg, C.W.; Bellin, M. Toward Human Models of Cardiorenal Syndrome in vitro. Front. Cardiovasc. Med. 2022, 9, 889553. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Demiray, A.; Afsar, B.; Covic, A.; Tapoi, L.; Ureche, C.; Ortiz, A. Role of Klotho in the Development of Essential Hypertension. Hypertension 2021, 77, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Tyurenkov, I.N.; Perfilova, V.N.; Nesterova, A.A.; Glinka, Y. Klotho Protein and Cardio-Vascular System. Biochemistry 2021, 86, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.; Totsuka, A.; Thompson, P.; Akatsuka, T.; Moritsugu, Y.; Feinstone, S.M. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996, 15, 4282–4296. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Bonventre, J.V.; Bailly, V.; Wei, H.; Hession, C.A.; Cate, R.L.; Sanicola, M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem. 1998, 273, 4135–4142. [Google Scholar] [CrossRef]
- McIntire, J.J.; Umetsu, S.E.; Akbari, O.; Potter, M.; Kuchroo, V.K.; Barsh, G.S.; Freeman, G.J.; Umetsu, D.T.; DeKruyff, R.H. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat. Immunol. 2001, 2, 1109–1116. [Google Scholar] [CrossRef]
- Li, Z.; Ju, Z.; Frieri, M. The T-cell immunoglobulin and mucin domain (Tim) gene family in asthma, allergy, and autoimmunity. Allergy Asthma Proc. 2013, 34, e21–e26. [Google Scholar] [CrossRef]
- Karmakova, T.; Sergeeva, N.S.; Kanukoev, K.Y.; Alekseev, B.Y.; Kaprin, A.D. Kidney Injury Molecule 1 (KIM-1): A Multifunctional Glycoprotein and Biological Marker (Review). Sovrem. Tekhnologii Med. 2021, 13, 64–78. [Google Scholar] [CrossRef]
- Medić, B.; Rovčanin, B.; Basta Jovanović, G.; Radojević-Škodrić, S.; Prostran, M. Kidney Injury Molecule-1 and Cardiovascular Diseases: From Basic Science to Clinical Practice. Biomed. Res. Int. 2015, 2015, 854070. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Masson, S.; Hillege, H.L.; Voors, A.A.; van Veldhuisen, D.J.; Rossignol, P.; Proietti, G.; Barbuzzi, S.; Nicolosi, G.L.; Tavazzi, L.; et al. Tubular Damage and Worsening Renal Function in Chronic Heart Failure. JACC Heart Fail. 2013, 1, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Lanza, O.; Savoia, C. New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. Int. J. Mol. Sci. 2023, 24, 5089. [Google Scholar] [CrossRef]
- Egli, P.; Aeschbacher, S.; Bossard, M.; Eggimann, L.; Blum, S.; Meyre, P.; Bargetzi, L.; Estis, J.; Todd, J.; Risch, M.; et al. Relationships of kidney injury molecule-1 with renal function and cardiovascular risk factors in the general population. Clin. Chim. Acta 2018, 478, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; El Moghrabi, S.; Messaoudi, S.; Nguyen Dinh Cat, A.; Hernandez-Diaz, I.; Alvarez de la Rosa, D.; Perret, C.; López Andrés, N.; Rossignol, P.; Zannad, F.; et al. Neutrophil Gelatinase-Associated Lipocalin Is a Novel Mineralocorticoid Target in the Cardiovascular System. Hypertension 2012, 59, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, H.; Chen, H.; Zhang, M.; Ma, Q. High expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney proximal tubules of diabetic rats. Adv. Med. Sci. 2015, 60, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Passov, A.; Petäjä, L.; Pihlajoki, M.; Salminen, U.-S.; Suojaranta, R.; Vento, A.; Andersson, S.; Pettilä, V.; Schramko, A.; Pesonen, E. The origin of plasma neutrophil gelatinase-associated lipocalin in cardiac surgery. BMC Nephrol. 2019, 20, 182. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta 2012, 1826, 129–169. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The Neutrophil Lipocalin NGAL Is a Bacteriostatic Agent that Interferes with Siderophore-Mediated Iron Acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Marakala, V. Neutrophil gelatinase-associated lipocalin (NGAL) in kidney injury–A systematic review. Clin. Chim. Acta 2022, 536, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Di Grande, A.; Giuffrida, C.; Carpinteri, G.; Narbone, G.; Pirrone, G.; Di Mauro, A.; Calandra, S.; Noto, P.; Le Moli, C.; Alongi, B.; et al. Neutrophil gelatinase-associated lipocalin: A novel biomarker for the early diagnosis of acute kidney injury in the emergency department. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 197–200. [Google Scholar]
- Parikh, C.R.; Devarajan, P. New biomarkers of acute kidney injury. Crit. Care Med. 2008, 36, S159–S165. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Jung, H.; Kwak, K.H.; Yi, S.J.; Lim, J.A.; Park, S.H.; Park, J.-M.; Kim, S.; Jee, D.-L.; Lim, D.G. Inhibition of Oxidative Stress in Renal Ischemia-Reperfusion Injury. Anesth. Analg. 2017, 124, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A Cell-Surface Receptor for Lipocalin 24p3 Selectively Mediates Apoptosis and Iron Uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Noel, S.; Kurzhagen, J.T.; Sadasivam, M.; Pierorazio, P.M.; Arend, L.J.; Hamad, A.R.; Rabb, H. CD4+ T Cell–Derived NGAL Modifies the Outcome of Ischemic Acute Kidney Injury. J. Immunol. 2020, 204, 586–595. [Google Scholar] [CrossRef]
- Jung, M.; Sola, A.; Hughes, J.; Kluth, D.C.; Vinuesa, E.; Viñas, J.L.; Pérez-Ladaga, A.; Hotter, G. Infusion of IL-10–expressing cells protects against renal ischem—Ia through induction of lipocalin-2. Kidney Int. 2012, 81, 969–982. [Google Scholar]
- Angelini, A.; Castellani, C.; Virzì, G.M.; Fedrigo, M.; Thiene, G.; Valente, M.; Ronco, C.; Vescovo, G. The Role of Congestion in Cardiorenal Syndrome Type 2: New Pathophysiological Insights into an Experimental Model of Heart Failure. Cardiorenal Med. 2015, 6, 61–72. [Google Scholar] [CrossRef]
- Vescovo, G.; Castellani, C.; Fedrigo, M.; Virzì, G.M.; Vescovo, G.M.; Tavano, R.; Pozzobon, M.; Angelini, A. Stem cells transplantation positively modulates the heart-kidney cross talk in cardiorenal syndrome type II. Int. J. Cardiol. 2019, 275, 136–144. [Google Scholar] [CrossRef]
- Buonafine, M.; Martinez-Martinez, E.; Jaisser, F. More than a simple biomarker: The role of NGAL in cardiovascular and renal diseases. Clin. Sci. 2018, 132, 909–923. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Xaplanteris, P.; Aboyans, V.; Brodmann, M.; Cífková, R.; Cosentino, F.; De Carlo, M.; Gallino, A.; Landmesser, U.; Laurent, S.; et al. The role of vascular biomarkers for primary and secondary prevention. A position paper from the European Society of Cardiology Working Group on peripheral circulation: Endorsed by the Association for Research into Arterial Structure and Physiology (ARTERY) Society. Atherosclerosis 2015, 241, 507–532. [Google Scholar] [PubMed]
- van Deursen, V.M.; Damman, K.; Voors, A.A.; van der Wal, M.H.; Jaarsma, T.; van Veldhuisen, D.J.; Hillege, H.L. Prognostic value of plasma neutrophil gelatinase-associated lipocalin for mortality in patients with heart failure. Circ. Heart Fail. 2014, 7, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, M.; Takahashi, M.; Doi, K.; Myojo, M.; Kiyosue, A.; Ando, J.; Hirata, Y.; Komuro, I. Serum neutrophil gelatinase-associated lipocalin concentration reflects severity of coronary artery disease in patients without heart failure and chronic kidney disease. Heart Vessel. 2016, 31, 1595–1602. [Google Scholar] [CrossRef]
- Martínez-Martínez, E.; Buonafine, M.; Boukhalfa, I.; Ibarrola, J.; Fernández-Celis, A.; Kolkhof, P.; Rossignol, P.; Girerd, N.; Mulder, P.; López-Andrés, N.; et al. Aldosterone Target NGAL (Neutrophil Gelatinase-Associated Lipocalin) Is Involved in Cardiac Remodeling After Myocardial Infarction Through NFκB Pathway. Hypertension 2017, 70, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martínez-Martínez, E.; Amador, C.; Gravez, B.; Ibarrola, J.; Fernández-Celis, A.; El Moghrabi, S.; Rossignol, P.; López-Andrés, N.; Jaisser, F. Neutrophil Gelatinase-Associated Lipocalin from immune cells is mandatory for aldosterone-induced cardiac remodeling and inflammation. J. Mol. Cell. Cardiol. 2018, 115, 32–38. [Google Scholar] [CrossRef]
- Miller, L.M.; Rifkin, D.; Lee, A.K.; Kurella Tamura, M.; Pajewski, N.M.; Weiner, D.E.; Al-Rousan, T.; Shlipak, M.; Ix, J.H. Association of Urine Biomarkers of Kidney Tubule Injury and Dysfunction With Frailty Index and Cognitive Function in Persons With CKD in SPRINT. Am. J. Kidney Dis. 2021, 78, 530–540. [Google Scholar] [CrossRef]
- Kanai, A.J.; Konieczko, E.M.; Bennett, R.G.; Samuel, C.S.; Royce, S.G. Relaxin and fibrosis: Emerging targets, challenges, and future directions. Mol. Cell. Endocrinol. 2019, 487, 66–74. [Google Scholar] [CrossRef]
- Bathgate, R.A.D.; Halls, M.L.; Westhuizen, E.T.v.d.; Callander, G.E.; Kocan, M.; Summers, R.J. Relaxin Family Peptides and Their Receptors. Physiol. Rev. 2013, 93, 405–480. [Google Scholar] [CrossRef]
- Bani, D. Relaxin: A pleiotropic hormone. General. Pharmacol. Vasc. Syst. 1997, 28, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.M.; Chew, E.G.Y.; Zhao, C.; Bathgate, R.A.D.; Hewitson, T.D.; Samuel, C.S. Relaxin Signals through a RXFP1-pERK-nNOS-NO-cGMP-Dependent Pathway to Up-Regulate Matrix Metalloproteinases: The Additional Involvement of iNOS. PLoS ONE 2012, 7, e42714. [Google Scholar] [CrossRef] [PubMed]
- Cernaro, V.; Lacquaniti, A.; Lupica, R.; Buemi, A.; Trimboli, D.; Giorgianni, G.; Bolignano, D.; Buemi, M. Relaxin: New pathophysiological aspects and pharmacological perspectives for an old protein. Med. Res. Rev. 2014, 34, 77–105. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Bathgate, R.A.; Sutton, S.W.; Dschietzig, T.B.; Summers, R.J. International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1-4, the receptors for relaxin family peptides. Med. Res. Rev. 2015, 34, 77–105. [Google Scholar] [CrossRef] [PubMed]
- Sassoli, C.; Nistri, S.; Chellini, F.; Bani, D. Human Recombinant Relaxin (Serelaxin) as Anti-fibrotic Agent: Pharmacology, Limitations and Actual Perspectives. Curr. Mol. Med. 2022, 22, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Pintalhao, M.; Castro-Chaves, P.; Vasques-Novoa, F.; Gonçalves, F.; Mendonça, L.; Fontes-Carvalho, R.; Lourenço, P.; Almeida, P.; Leite-Moreira, A.; Bettencourt, P. Relaxin serum levels in acute heart failure are associated with pulmonary hypertension and right heart overload. Eur. J. Heart Fail. 2017, 19, 218–225. [Google Scholar] [CrossRef]
- Henry, B.L.; Gabris, B.; Li, Q.; Martin, B.; Giannini, M.; Parikh, A.; Patel, D.; Haney, J.; Schwartzman, D.S.; Shroff, S.G.; et al. Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na+ channels. Heart Rhythm 2016, 13, 983–991. [Google Scholar] [CrossRef]
- Ureche, C.; Nedelcu, A.-E.; Sascău, R.A.; Stătescu, C.; Kanbay, M.; Covic, A. Role of collagen turnover biomarkers in the noninvasive assessment of myocardial fibrosis: An update. Biomark. Med. 2020, 14, 1265–1275. [Google Scholar] [CrossRef]
- Demir, S.; Ede, H.; Kaplan, M.; Yavuz, F.; Yucel, C.; Kurt, I.H. The novel diagnostic marker in low-LVEF heart failure patients. Bratisl. Lek. Listy 2018, 119, 421–424. [Google Scholar] [CrossRef]
- Salib, M.; Girerd, S.; Girerd, N.; März, W.; Scharnagl, H.; Massy, Z.A.; Leroy, C.; Duarte, K.; Holdaas, H.; Jardine, A.G.; et al. Serum markers of fibrosis, cardiovascular and all-cause mortality in hemodialysis patients: The AURORA trial. Clin. Res. Cardiol. 2022, 111, 614–626. [Google Scholar] [CrossRef]
- Ureche, C.; Dodi, G.; Covic, A.; Nedelcu, A.; Volovăț, S.R.; Sascău, R.A.; Stătescu, C.; Covic, A. Connection between Cardiac Fibrosis Biomarkers and Echocardiography Parameters in Advanced Chronic Kidney Disease Patients. J. Clin. Med. 2023, 12, 3003. [Google Scholar] [CrossRef] [PubMed]
- Löfsjögård, J.; Kahan, T.; Díez, J.; López, B.; González, A.; Ravassa, S.; Mejhert, M.; Edner, M.; Persson, H. Usefulness of Collagen Carboxy-Terminal Propeptide and Telopeptide to Predict Disturbances of Long-Term Mortality in Patients ≥60 Years With Heart Failure and Reduced Ejection Fraction. Am. J. Cardiol. 2017, 119, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Duprez, D.A.; Gross, M.D.; Kizer, J.R.; Ix, J.H.; Hundley, W.G.; Jacobs, D.R.J. Predictive Value of Collagen Biomarkers for Heart Failure with and without Preserved Ejection Fraction: MESA (Multi-Ethnic Study of Atherosclerosis). J. Am. Heart Assoc. 2018, 7, e007885. [Google Scholar] [CrossRef] [PubMed]
- Kanoupakis, E.M.; Manios, E.G.; Kallergis, E.M.; Mavrakis, H.E.; Goudis, C.A.; Saloustros, I.G.; Milathianaki, M.E.; Chlouverakis, G.I.; Vardas, P.E. Serum markers of collagen turnover predict future shocks in implantable cardioverter-defibrillator recipients with dilated cardiomyopathy on optimal treatment. J. Am. Coll. Cardiol. 2010, 55, 2753–2759. [Google Scholar] [CrossRef] [PubMed]
- Eschalier, R.; Fertin, M.; Fay, R.; Bauters, C.; Zannad, F.; Pinet, F.; Rossignol, P. Extracellular matrix turnover biomarkers predict long-term left ventricular remodeling after myocardial infarction: Insights from the REVE-2 study. Circ. Heart Fail. 2013, 6, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Ghoul, B.E.; Squalli, T.; Servais, A.; Elie, C.; Meas-Yedid, V.; Trivint, C.; Vanmassenhove, J.; Grünfeld, J.P.; Olivo-Marin, J.C.; Thervet, E.; et al. Urinary procollagen III aminoterminal propeptide (PIIINP): A fibrotest for the nephrologist. Clin. J. Am. Soc. Nephrol. 2010, 5, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ureche, C.; Dodi, G.; Șerban, A.M.; Covic, A.S.; Voroneanu, L.; Hogaș, S.; Sascău, R.A.; Stătescu, C.; Covic, A. Predictive Value of Collagen Biomarkers in Advanced Chronic Kidney Disease Patients. Biomolecules 2023, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Zhao, L.; Reese-Petersen, A.L.; Cohen, J.B.; Genovese, F.; Richards, A.M.; Doughty, R.N.; Díez, J.; González, A.; Querejeta, R.; et al. Endotrophin, a Collagen VI Formation-Derived Peptide, in Heart Failure. NEJM Evid. 2022, 1, EVIDoa2200091. [Google Scholar] [CrossRef]
- Reese-Petersen, A.L.; Genovese, F.; Zhao, L.; Banks, G.; Gordon, D.A.; Karsdal, M.A. Endotrophin, a fibroblast matrikine, may be a driver of fibroblast activation in fibro-inflammatory diseases. Front. Mol. Biosci. 2023, 10, 1228232. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Z. Effect and Regulation of the NLRP3 Inflammasome During Renal Fibrosis. Front. Cell Dev. Biol. 2019, 7, 379. [Google Scholar] [CrossRef]
- Su, X.; Liu, B.; Wang, S.; Wang, Y.; Zhang, Z.; Zhou, H.; Li, F. NLRP3 inflammasome: A potential therapeutic target to minimize renal ischemia/reperfusion injury during transplantation. Transpl. Immunol. 2022, 75, 101718. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, X.; Chen, S.; Guo, X. The AIM2 inflammasome: A novel biomarker and target in cardiovascular disease. Pharmacol. Res. 2022, 186, 106533. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Buckley, L.F.; Potere, N.; Di Nisio, M.; Biondi-Zoccai, G.; Van Tassell, B.W.; Abbate, A. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 2022, 236, 108053. [Google Scholar] [CrossRef] [PubMed]
- Clarisse, D.; Deng, L.; de Bosscher, K.; Lother, A. Approaches towards tissue-selective pharmacology of the mineralocorticoid receptor. Br. J. Pharmacol. 2022, 179, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Zhou, M.; Chen, W.; Li, D.; Wang, Z.; Hornsperger, B.; Aebi, J.D.; Märki, H.-P.; Kuhn, B.; et al. Discovery of 3-Pyridyl Isoindolin-1-one Derivatives as Potent, Selective, and Orally Active Aldosterone Synthase (CYP11B2) Inhibitors. J. Med. Chem. 2020, 63, 6876–6897. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Bayer. A Study to Learn How Well the Treatment Combination of Finerenone and Empagliflozin Works and How Safe it is Compared to Each Treatment Alone in Adult Participants with Long-Term Kidney Disease (Chronic Kidney Disease) and Type 2 Diabetes. Available online: https://classic.clinicaltrials.gov/show/NCT05254002:2022 (accessed on 13 February 2024).
- AstraZeneca. Efficacy, Safety and Tolerability of AZD9977 and Dapagliflozin in Participants with Heart Failure and Chronic Kidney Disease. Available online: https://classic.clinicaltrials.gov/show/NCT04595370:2021 (accessed on 13 February 2024).
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef]
- Bani, D. Recombinant human H2 relaxin (serelaxin) as a cardiovascular drug: Aiming at the right target. Drug Discov. Today 2020, 25, 1239–1244. [Google Scholar] [CrossRef]
- Giam, B.; Chu, P.-Y.; Kuruppu, S.; Smith, A.I.; Horlock, D.; Murali, A.; Kiriazis, H.; Du, X.-J.; Kaye, D.M.; Rajapakse, N.W. Serelaxin attenuates renal inflammation and fibrosis in a mouse model of dilated cardiomyopathy. Exp. Physiol. 2018, 103, 1593–1602. [Google Scholar] [CrossRef]
- Wilhelmi, T.; Xu, X.; Tan, X.; Hulshoff, M.S.; Maamari, S.; Sossalla, S.; Zeisberg, M.; Zeisberg, E.M. Serelaxin alleviates cardiac fibrosis through inhibiting endothelial-to-mesenchymal transition via RXFP1. Theranostics 2020, 10, 3905–3924. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Ponikowski, P.; Unemori, E.; Voors, A.A.; Adams, K.F.J.; et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): A randomised, placebo-controlled trial. Lancet 2013, 381, 29–39. [Google Scholar] [CrossRef]
- Metra, M.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Voors, A.A.; et al. Effects of Serelaxin in Patients with Acute Heart Failure. N. Engl. J. Med. 2019, 381, 716–726. [Google Scholar] [CrossRef]
- Chow, B.S.M.; Kocan, M.; Shen, M.; Wang, Y.; Han, L.; Chew, J.Y.; Wang, C.; Bosnyak, S.; Mirabito-Colafella, K.M.; Barsha, G.; et al. AT1R-AT2R-RXFP1 Functional Crosstalk in Myofibroblasts: Impact on the Therapeutic Targeting of Renal and Cardiac Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 2191–2207. [Google Scholar] [CrossRef]
- Corden, B.; Adami, E.; Sweeney, M.; Schafer, S.; Cook, S.A. IL-11 in cardiac and renal fibrosis: Late to the party but a central player. Br. J. Pharmacol. 2020, 177, 1695–1708. [Google Scholar] [CrossRef]
- Ng, B.A.-O.; Dong, J.A.-O.; D’Agostino, G.A.-O.; Viswanathan, S.; Widjaja, A.A.-O.; Lim, W.A.-O.X.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.A.-O.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D’Agostino, G.A.; Ng, B.; Lim, W.W.; Tan, J.; Paleja, B.S.; et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e14. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, A.A.; Shekeran, S.G.; Adami, E.; Ting, J.G.W.; Tan, J.; Viswanathan, S.; Lim, S.Y.; Tan, P.H.; Hübner, N.; Coffman, T.; et al. A Neutralizing IL-11 Antibody Improves Renal Function and Increases Lifespan in a Mouse Model of Alport Syndrome. J. Am. Soc. Nephrol. 2022, 33, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, A.A.; Viswanathan, S.; Shekeran, S.G.; Adami, E.; Lim, W.W.; Chothani, S.; Tan, J.; Goh, J.W.T.; Chen, H.M.; Lim, S.Y.; et al. Targeting endogenous kidney regeneration using anti-IL11 therapy in acute and chronic models of kidney disease. Nat. Commun. 2022, 13, 7497. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Machu, J.L.; Girerd, N.; Jaisser, F.; Thum, T.; Butler, J.; González, A.; Diez, J.; Heymans, S.; McDonald, K.; et al. Rationale of the FIBROTARGETS study designed to identify novel biomarkers of myocardial fibrosis. ESC Heart Fail. 2018, 5, 139–148. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiuariu, T.; Șalaru, D.; Ureche, C.; Vasiliu, L.; Lupu, A.; Lupu, V.V.; Șerban, A.M.; Zăvoi, A.; Benchea, L.C.; Clement, A.; et al. Cardiac and Renal Fibrosis, the Silent Killer in the Cardiovascular Continuum: An Up-to-Date. J. Cardiovasc. Dev. Dis. 2024, 11, 62. https://doi.org/10.3390/jcdd11020062
Chiuariu T, Șalaru D, Ureche C, Vasiliu L, Lupu A, Lupu VV, Șerban AM, Zăvoi A, Benchea LC, Clement A, et al. Cardiac and Renal Fibrosis, the Silent Killer in the Cardiovascular Continuum: An Up-to-Date. Journal of Cardiovascular Development and Disease. 2024; 11(2):62. https://doi.org/10.3390/jcdd11020062
Chicago/Turabian StyleChiuariu, Traian, Delia Șalaru, Carina Ureche, Laura Vasiliu, Ancuta Lupu, Vasile Valeriu Lupu, Adela Mihaela Șerban, Alexandra Zăvoi, Laura Catalina Benchea, Alexandra Clement, and et al. 2024. "Cardiac and Renal Fibrosis, the Silent Killer in the Cardiovascular Continuum: An Up-to-Date" Journal of Cardiovascular Development and Disease 11, no. 2: 62. https://doi.org/10.3390/jcdd11020062
APA StyleChiuariu, T., Șalaru, D., Ureche, C., Vasiliu, L., Lupu, A., Lupu, V. V., Șerban, A. M., Zăvoi, A., Benchea, L. C., Clement, A., Tudurachi, B.-S., Sascău, R. A., & Stătescu, C. (2024). Cardiac and Renal Fibrosis, the Silent Killer in the Cardiovascular Continuum: An Up-to-Date. Journal of Cardiovascular Development and Disease, 11(2), 62. https://doi.org/10.3390/jcdd11020062