
Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect?


 ,
,  and
and 
Abstract
1. Introduction
2. Neutrophil Extracellular Traps (NETs)
2.1. Definition and Formation
2.2. Triggers
2.3. NETs Content and Quantification
3. Correlation of NETs to Atherosclerosis and Implication in Its Pathogenesis
3.1. Atherosclerosis, Immunity and Inflammation
3.2. Complications Leading to ASCVD Events
3.3. The Role of NETs in Atherosclerosis
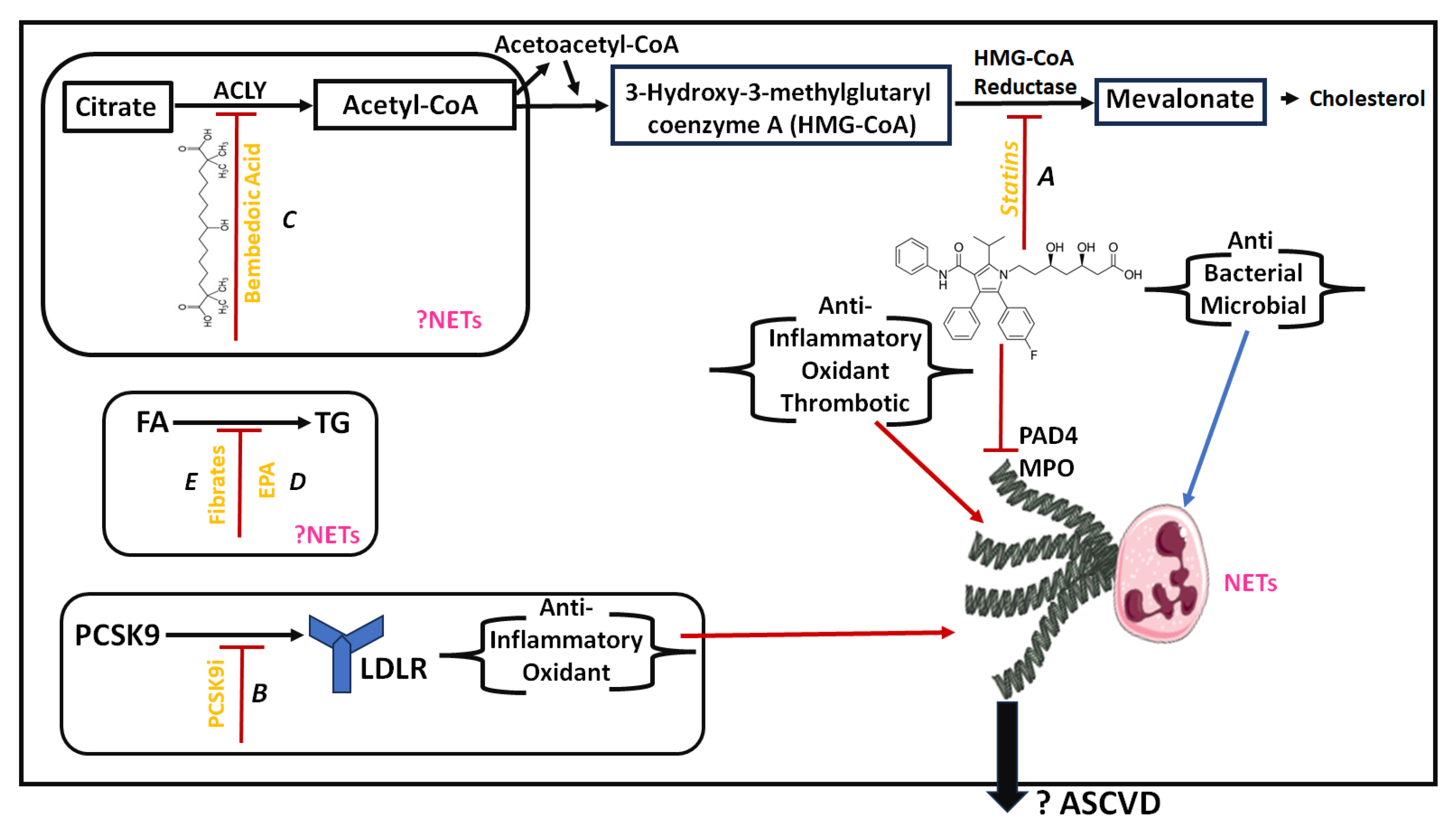
4. Lipid-Lowering Treatment and NETs
4.1. Current Knowledge on Lipid-Lowering Treatment
4.2. Documented Effects of Lipid-Lowering Treatment on NETs and NETosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Pan, Q.; Chen, C.; Yang, Y.J. Top Five Stories of the Cellular Landscape and Therapies of Atherosclerosis: Current Knowledge and Future Perspectives. Curr. Med. Sci. 2023, 2023, 1–27. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Shimony, A.; Zahger, D.; Gilutz, H.; Goldstein, H.; Orlov, G.; Merkin, M.; Shalev, A.; Ilia, R.; Douvdevani, A. Cell free DNA detected by a novel method in acute ST-elevation myocardial infarction patients. Acute Card. Care 2010, 12, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Fan, M.; Jing, R.; Wang, H.; Qin, J.; Sheng, H.; Wang, Y.; Wu, X.; Zhang, L.; Zhu, J.; et al. Cell-free circulating DNA: A new biomarker for the acute coronary syndrome. Cardiology 2013, 124, 76–84. [Google Scholar] [CrossRef]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Alsac, J.-M.; Journe, C.; Louedec, L.; Castier, Y.; Bonnaure-Mallet, M.; Ruimy, R.; Rossignol, P.; Bouchard, P.; Michel, J.-B.; et al. Porphyromonas gingivalis Participates in Pathogenesis of Human Abdominal Aortic Aneurysm by Neutrophil Activation. Proof of Concept in Rats. PLoS ONE 2011, 6, e18679. [Google Scholar] [CrossRef]
- Yan, H.; Zhou, H.F.; Akk, A.; Hu, Y.; Springer, L.E.; Ennis, T.L.; Pham, C.T.N. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1660–1669. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines, Health Research Alliance manuscript submission. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Hong, Y.M. Atherosclerotic Cardiovascular Disease Beginning in Childhood. Korean Circ. J. 2010, 40, 1. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; Van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Tokgözoǧlu, L.; Libby, P. The dawn of a new era of targeted lipid-lowering therapies. Eur. Heart J. 2022, 43, 3198–3208. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Moschonas, I.C.; Tselepis, A.D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis 2019, 288, 9–16. [Google Scholar] [CrossRef]
- Welin, A.; Amirbeagi, F.; Christenson, K.; Björkman, L.; Björnsdottir, H.; Forsman, H.; Dahlgren, C.; Karlsson, A.; Bylund, J. The Human Neutrophil Subsets Defined by the Presence or Absence of OLFM4 Both Transmigrate into Tissue In Vivo and Give Rise to Distinct NETs In Vitro. PLoS ONE 2013, 8, e69575. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Ger, R.K.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef]
- Abdallah, D.S.A.; Lin, C.; Ball, C.J.; King, M.R.; Duhamel, G.E.; Denkers, E.Y. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect. Immun. 2012, 80, 768–777. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Pieterse, E.; Jeremic, I.; Czegley, C.; Weidner, D.; Biermann, M.H.C.; Veissi, S.; Maueröder, C.; Schauer, C.; Bilyy, R.; Dumych, T.; et al. Blood-borne phagocytes internalize urate microaggregates and prevent intravascular NETosis by urate crystals. Sci. Rep. 2016, 6, 38229. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Thålin, C.; Lundström, S.; Seignez, C.; Daleskog, M.; Lundström, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallén, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Cate, H.T.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state, Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef]
- Obama, T.; Itabe, H. Neutrophils as a Novel Target of Modified Low-Density Lipoproteins and an Accelerator of Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 8312. [Google Scholar] [CrossRef]
- Masuda, S.; Nakazawa, D.; Shida, H.; Miyoshi, A.; Kusunoki, Y.; Tomaru, U.; Ishizu, A. NETosis markers: Quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016, 459, 89–93. [Google Scholar] [CrossRef]
- Pantazi, D.; Tellis, C.; Tselepis, A.D. Oxidized phospholipids and lipoprotein-associated phospholipase A2 (Lp-PLA2) in atherosclerotic cardiovascular disease: An update. Biofactors 2022, 48, 1257–1270. [Google Scholar] [CrossRef]
- Hansen, S.E.J.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Low-Grade Inflammation in the Association between Mild-to-Moderate Hypertriglyceridemia and Risk of Acute Pancreatitis: A Study of More Than 115000 Individuals from the General Population. Clin. Chem. 2019, 65, 321–332. [Google Scholar] [CrossRef]
- Varbo, A.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation 2013, 128, 1298–1309. [Google Scholar] [CrossRef]
- Ridker, P.M. A Test in Context: High-Sensitivity C-Reactive Protein. J. Am. Coll. Cardiol. 2016, 67, 712–723. [Google Scholar] [CrossRef]
- Xiao, L.; Harrison, D.G. Inflammation in Hypertension. Can. J. Cardiol. 2020, 36, 635–647. [Google Scholar] [CrossRef]
- Ross, R.; Neeland, I.J.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; Cuevas, A.; Hu, F.B.; et al. Waist circumference as a vital sign in clinical practice: A Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat. Rev. Endocrinol. 2020, 16, 177–189. [Google Scholar] [CrossRef]
- Giovenzana, A.; Carnovale, D.; Phillips, B.; Petrelli, A.; Giannoukakis, N. Neutrophils and their role in the aetiopathogenesis of type 1 and type 2 diabetes. Diabetes. Metab. Res. Rev. 2021, 38, e3483. [Google Scholar] [CrossRef]
- Libby, P.; Hansson, G.K. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1594–1607. [Google Scholar] [CrossRef]
- Paulson, K.E.; Zhu, S.N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef]
- Lim, H.Y.; Lim, S.Y.; Tan, C.K.; Thiam, C.H.; Goh, C.C.; Carbajo, D.; Chew, S.H.S.; See, P.; Chakarov, S.; Wang, X.N.; et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018, 49, 326–341.e7. [Google Scholar] [CrossRef]
- Owsiany, K.M.; Alencar, G.F.; Owens, G.K. Revealing the origins of foam cells in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 836. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; La Bastide-Van Gemert, S.; Wang, N. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Paulin, N.; Viola, J.R.; Maas, S.L.; De Jong, R.; Fernandes-Alnemri, T.; Weber, C.; Drechsler, M.; Döring, Y.; Soehnlein, O. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation 2018, 138, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Collagenases and cracks in the plaque. J. Clin. Investig. 2013, 123, 3201–3203. [Google Scholar] [CrossRef]
- Davies, M.J. Stability and instability: Two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation 1996, 94, 2013–2020. [Google Scholar] [CrossRef]
- Douglas, P.S.; Hoffmann, U.; Patel, M.R.; Mark, D.B.; Al-Khalidi, H.R.; Cavanaugh, B.; Cole, J.; Dolor, R.J.; Fordyce, C.B.; Huang, M.; et al. Outcomes of anatomical versus functional testing for coronary artery disease. N. Engl. J. Med. 2015, 372, 1291–1300. [Google Scholar] [CrossRef]
- Coronary CT Angiography and 5-Year Risk of Myocardial Infarction. N. Engl. J. Med. 2018, 379, 924–933. [CrossRef]
- Stone, G.W.; Maehara, A.; Lansky, A.J.; de Bruyne, B.; Cristea, E.; Mintz, G.S.; Mehran, R.; McPherson, J.; Farhat, N.; Marso, S.P.; et al. A prospective natural-history study of coronary atherosclerosis. N. Engl. J. Med. 2011, 364, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Franck, G.; Even, G.; Gautier, A.; Salinas, M.; Loste, A.; Procopio, E.; Gaston, A.T.; Morvan, M.; Dupont, S.; Deschildre, C.; et al. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur. Heart J. 2019, 40, 928–937. [Google Scholar] [CrossRef]
- Libby, P. Once more unto the breach: Endothelial permeability and atherogenesis. Eur. Heart J. 2019, 40, 938–940. [Google Scholar] [CrossRef]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021, 117, 2652–2663. [Google Scholar] [CrossRef]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; Van Der Wal, A.C.; Pabittei, D.R.; Mackaaij, C.; Van Leeuwen, M.B.; Li, X.; De Boer, O.J. Neutrophil Extracellular Traps Participate in All Different Types of Thrombotic and Haemorrhagic Complications of Coronary Atherosclerosis. Thromb. Haemost. 2018, 118, 1078–1087. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947. [Google Scholar] [CrossRef]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Hemmers, S.; Arandjelovic, S.; Corr, M.; Mowen, K.A. PAD4 is not essential for disease in the K/BxN murine autoantibody-mediated model of arthritis. Arthritis Res. Ther. 2012, 14, R104. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Neutrophil extracellular traps: A new source of tissue factor in atherothrombosis. Eur. Heart J. 2015, 36, 1364–1366. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2020, 18, 1528–1544. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.; Shinagawa, H.; Hofmann, U.; Ertl, G.; Frantz, S. Acute DNase1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin. Basic Res. Cardiol. 2015, 110, 15. [Google Scholar] [CrossRef]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef] [PubMed]
- Kessinger, C.W.; Kim, J.W.; Henke, P.K.; Thompson, B.; McCarthy, J.R.; Hara, T.; Sillesen, M.; Margey, R.J.P.; Libby, P.; Weissleder, R.; et al. Statins improve the resolution of established murine venous thrombosis: Reductions in thrombus burden and vein wall scarring. PLoS ONE 2015, 10, e0116621. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghoul, W.M.; Kim, M.S.; Fazal, N.; Azim, A.C.; Ali, A. Evidence for simvastatin anti-inflammatory actions based on quantitative analyses of NETosis and other inflammation/oxidation markers. Results Immunol. 2014, 4, 14–22. [Google Scholar] [CrossRef]
- Chow, O.A.; Von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef]
- Henneck, T.; Mergani, A.; Clever, S.; Seidler, A.E.; Brogden, G.; Runft, S.; Baumgärtner, W.; Branitzki-Heinemann, K.; von Köckritz-Blickwede, M.V. Formation of Neutrophil Extracellular Traps by Reduction of Cellular Cholesterol Is Independent of Oxygen and HIF-1α. Int. J. Mol. Sci. 2022, 23, 3195. [Google Scholar] [CrossRef] [PubMed]
- Donkel, S.J.; Wolters, F.J.; Ikram, M.A.; de Maat, M.P.M. Circulating Myeloperoxidase (MPO)-DNA complexes as marker for Neutrophil Extracellular Traps (NETs) levels and the association with cardiovascular risk factors in the general population. PLoS ONE 2021, 16, e0253698. [Google Scholar] [CrossRef]
- Cholesterol Loading Induces Neutrophil Extracellular Traps, and Atorvastatin Attenuates This Effect—ACR Meeting Abstracts, (n.d.). Available online: https://acrabstracts.org/abstract/cholesterol-loading-induces-neutrophil-extracellular-traps-and-atorvastatin-attenuates-this-effect/ (accessed on 20 December 2023).
- Park, H.S.; Gu, J.Y.; Yoo, H.J.; Han, S.E.; Park, C.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Thrombin Generation Assay Detects Moderate-Intensity Statin-Induced Reduction of Hypercoagulability in Diabetes. Clin. Appl. Thromb. 2018, 24, 1095. [Google Scholar] [CrossRef]
- de Vries, J.J.; Autar, A.S.A.; van Dam-Nolen, D.H.K.; Donkel, S.J.; Kassem, M.; van der Kolk, A.G.; van Velzen, T.J.; Kooi, M.E.; Hendrikse, J.; Nederkoorn, P.J.; et al. Association between plaque vulnerability and neutrophil extracellular traps (NETs) levels: The Plaque at RISK study. PLoS ONE 2022, 17, e0269805. [Google Scholar] [CrossRef]
- Sapey, E.; Patel, J.M.; Greenwood, H.; Walton, G.M.; Grudzinska, F.; Parekh, D.; Mahida, R.Y.; Dancer, R.C.A.; Lugg, S.T.; Howells, P.A.; et al. Simvastatin improves neutrophil function and clinical outcomes in pneumonia a pilot randomized controlled clinical trial. Am. J. Respir. Crit. Care Med. 2019, 200, 1282–1293. [Google Scholar] [CrossRef]
- Chen, Y.R.; Xiang, X.D.; Sun, F.; Xiao, B.W.; Yan, M.Y.; Peng, B.; Liu, D. Simvastatin Reduces NETosis to Attenuate Severe Asthma by Inhibiting PAD4 Expression. Oxid. Med. Cell. Longev. 2023, 2023, 1493684. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Wang, J.; Guo, C.; Kleiman, K.; Meng, H.; Knight, J.S.; Eitzman, D.T. Proprotein convertase subtilisin/kexin type 9 (PCSK9) Deficiency is Protective against Venous Thrombosis in Mice. Sci. Rep. 2017, 7, 14360. [Google Scholar] [CrossRef]
- Yang, J.; Ma, X.; Niu, D.; Sun, Y.; Chai, X.; Deng, Y.; Wang, J.; Dong, J. PCSK9 inhibitors suppress oxidative stress and inflammation in atherosclerotic development by promoting macrophage autophagy. Am. J. Transl. Res. 2023, 15, 5129. [Google Scholar] [PubMed]
- Scicali, R.; Di Pino, A.; Ferrara, V.; Rabuazzo, A.M.; Purrello, F.; Piro, S. Effect of PCSK9 inhibitors on pulse wave velocity and monocyte-to-HDL-cholesterol ratio in familial hypercholesterolemia subjects: Results from a single-lipid-unit real-life setting. Acta Diabetol. 2021, 58, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Haghikia, A.; Leiter, L.A.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Stoekenbroek, R.; Kastelein, J.J.; Ray, K.K. Effect of inclisiran, the small-interfering RNA against proprotein convertase subtilisin/kexin type 9, on platelets, immune cells, and immunological biomarkers: A pre-specified analysis from ORION-1. Cardiovasc. Res. 2021, 117, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Ginsberg, H.N.; Choi, S.H. New, Novel Lipid-Lowering Agents for Reducing Cardiovascular Risk: Beyond Statins. Diabetes Metab. J. 2022, 46, 517. [Google Scholar] [CrossRef]

{kind=link}
| Authors | Year | Type of Study | Results |
|---|---|---|---|
| Chow et al. [85] | 2010 | Experimental | Mevastatin, lovastatin, simvastatin, fluvastatin enhance NETs production of human neutrophils in vitro. |
| Kessinger et al. [83] | 2015 | Experimental | Atorvastatin compared to PBS reduced levels of neutrophils and CitH3 in murine thrombi. |
| Al-Ghoul et al. [84] | 2014 | Experimental | Simvastatin exerted protective effect against inflammation and systemic NETosis post-thermal injury. |
| Liu et al. [88] | 2014 | Experimental | Pretreatment with atorvastatin alleviated the cholesterol-induced NETs production in vitro. |
| Park et al. [89] | 2018 | Clinical | NETosis biomarkers (NE, DNA–histone complexes, cell-free DNA) levels decreased non-significantly after 3 month treatment with moderate intensity statins in 25 diabetic patients. |
| Wang et al. [93] | 2017 | Experimental | NETs formation and leucocyte accumulation significantly reduced in PCSK9 -/- mice compared to wild-type controls. |
| De Vries et al. [90] | 2022 | Clinical | Attenuation of plasma NETosis components association with atherosclerotic plaque vulnerability index probably via effects in NETs levels or functions. |
| Henneck et al. [86] | 2022 | Experimental | Simvastatin and mevastatin trigger NETs formation in isolated neutrophils by depleting intracellular cholesterol independently from oxygen supply. |
| Sapey et al. [91] | 2019 | Clinical | Add-on high dose simvastatin versus placebo on 62 patients with community-acquired pneumonia with sepsis reduced NETosis on treatment day 4. |
| Chen et al. [92] | 2023 | Experimental | Simvastatin reduced NETs formation in bronchoalveolar lavage and lung tissue in a murine model of severe asthma. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamidis, P.S.; Pantazi, D.; Moschonas, I.C.; Liberopoulos, E.; Tselepis, A.D. Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect? J. Cardiovasc. Dev. Dis. 2024, 11, 72. https://doi.org/10.3390/jcdd11030072
Adamidis PS, Pantazi D, Moschonas IC, Liberopoulos E, Tselepis AD. Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect? Journal of Cardiovascular Development and Disease. 2024; 11(3):72. https://doi.org/10.3390/jcdd11030072
Chicago/Turabian StyleAdamidis, Petros Spyridonas, Despoina Pantazi, Iraklis C. Moschonas, Evangelos Liberopoulos, and Alexandros D. Tselepis. 2024. "Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect?" Journal of Cardiovascular Development and Disease 11, no. 3: 72. https://doi.org/10.3390/jcdd11030072
APA StyleAdamidis, P. S., Pantazi, D., Moschonas, I. C., Liberopoulos, E., & Tselepis, A. D. (2024). Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect? Journal of Cardiovascular Development and Disease, 11(3), 72. https://doi.org/10.3390/jcdd11030072