
Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity


Abstract
1. Introduction
2. Materials and Methods
3. Risk Calculations in Statin Trials
Reassessing Landmark Statin Trial Findings
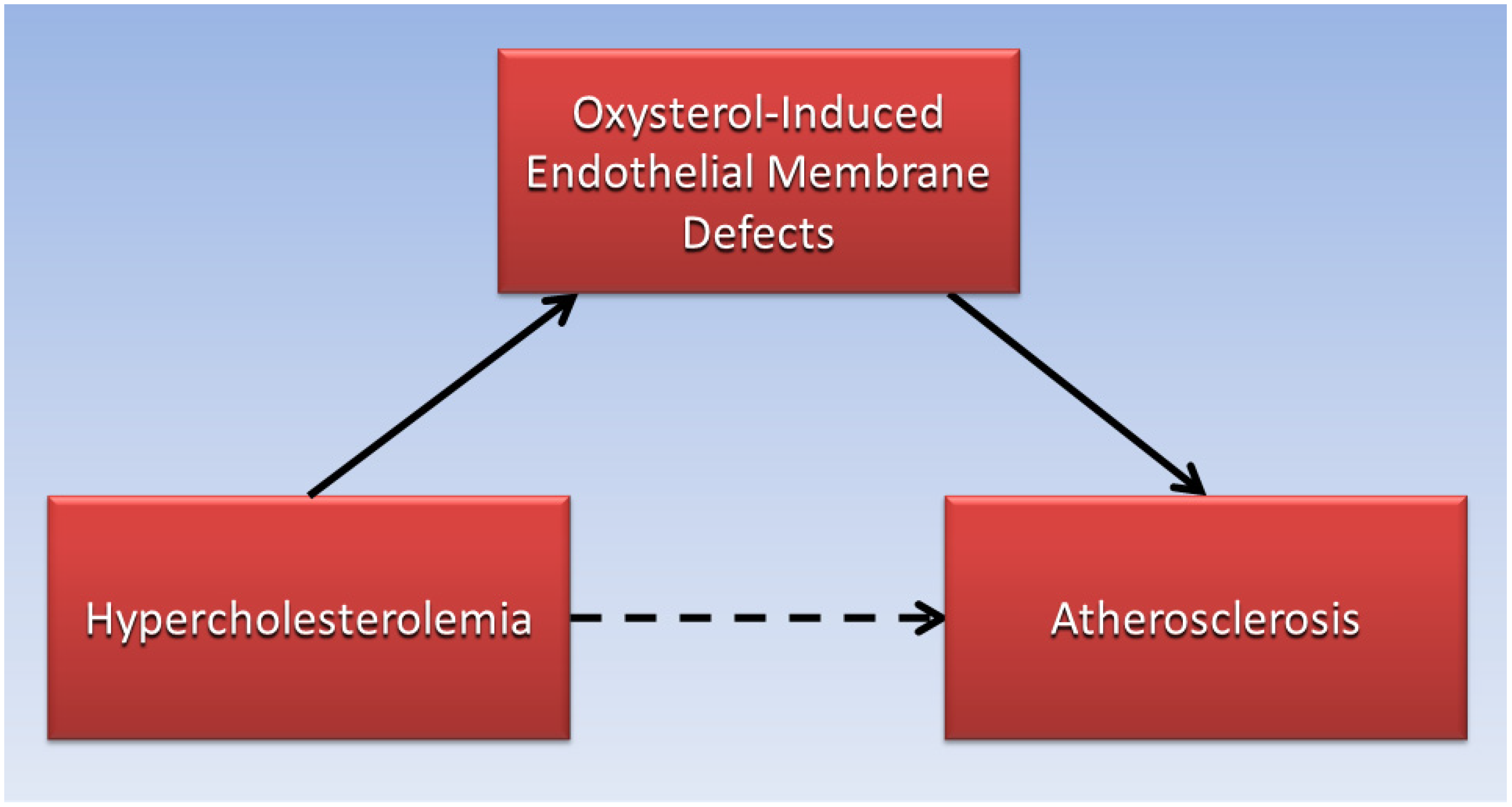
4. Cholesterol and Oxysterols
5. Statins and Increased Cancer Risk
6. Statins’ Anticancer Effect and Confounding by Indication
7. Tumorigenesis and Phosphate Toxicity
8. Dietary Patterns and Confounding by Indication
9. Statin-Induced Rhabdomyolysis and Phosphate Toxicity
9.1. Diabetes
9.2. Parkinson’s Disease
9.3. Cardiomyopathy
9.4. Kidney Disease
9.5. Bone Fracture, Cataract, and Coronary Artery Calcification
9.6. Vitamin D Deficiency
9.7. Periodontal Disease
9.8. Osteoarthritis
10. Summary of Statin-Cancer Risk Factors
11. Future Research
12. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- How Statin Drugs Protect the Heart. 2023. Available online: https://www.hopkinsmedicine.org/health/wellness-and-prevention/how-statin-drugs-protect-the-heart (accessed on 22 June 2023).
- Guadamuz, J.S.; Shooshtari, A.; Qato, D.M. Global, regional and national trends in statin utilisation in high-income and low/middle-income countries, 2015–2020. BMJ Open 2022, 12, e061350. [Google Scholar] [CrossRef] [PubMed]
- Tilija Pun, N.; Jeong, C.H. Statin as a Potential Chemotherapeutic Agent: Current Updates as a Monotherapy, Combination Therapy, and Treatment for Anti-Cancer Drug Resistance. Pharmaceuticals 2021, 14, 470. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Y.; Fard, J.K.; Ghafoor, D.; Eid, A.H.; Sahebkar, A. Paradoxical effects of statins on endothelial and cancer cells: The impact of concentrations. Cancer Cell Int. 2023, 23, 43. [Google Scholar] [CrossRef]
- Göbel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond—The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta (BBA)-Rev. Cancer. 2020, 1873, 188351. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Mei, Z.; Liang, M.; Li, L.; Zhang, Y.; Wang, Q.; Yang, W. Effects of statins on cancer mortality and progression: A systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int. J. Cancer 2017, 140, 1068–1081. [Google Scholar] [CrossRef]
- Tamburrino, D.; Crippa, S.; Partelli, S.; Archibugi, L.; Arcidiacono, P.G.; Falconi, M.; Capurso, G. Statin use improves survival in patients with pancreatic ductal adenocarcinoma: A meta-analysis. Dig. Liver Dis. 2020, 52, 392–399. [Google Scholar] [CrossRef]
- Majidi, A.; Na, R.; Jordan, S.J.; De Fazio, A.; Webb, P.M. Statin use and survival following a diagnosis of ovarian cancer: A prospective observational study. Int. J. Cancer 2021, 148, 1608–1615. [Google Scholar] [CrossRef]
- Islam, M.M.; Poly, T.N.; Walther, B.A.; Yang, H.C.; Jack Li, Y.C. Statin Use and the Risk of Hepatocellular Carcinoma: A Meta-Analysis of Observational Studies. Cancers 2020, 12, 671. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Reimers, M.S.; Bastiaannet, E.; Jacobs, R.J.; van Eijk, R.; Zanders, M.M.; Herings, R.M.; van Herk-Sukel, M.P.; Kodach, L.L.; van Wezel, T.; et al. Statin Use After Diagnosis of Colon Cancer and Patient Survival. Gastroenterology 2017, 153, 470–479.e4. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Johnson, M.L.; Hachem, C.; Morgana, R.O. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 2009, 136, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Beckwitt, C.H.; Clark, A.M.; Ma, B.; Whaley, D.; Oltvai, Z.N.; Wells, A. Statins attenuate outgrowth of breast cancer metastases. Br. J. Cancer 2018, 119, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, W.; Wang, J.; Xie, L.; Li, T.; He, Y.; Deng, Y.; Peng, Q.; Li, S.; Qin, X. Association between statin use and colorectal cancer risk: A meta-analysis of 42 studies. Cancer Causes Control 2014, 25, 237–249. [Google Scholar] [CrossRef]
- Bonovas, S.; Filioussi, K.; Tsavaris, N.; Sitaras, N.M. Use of statins and breast cancer: A meta-analysis of seven randomized clinical trials and nine observational studies. J. Clin. Oncol. 2005, 23, 8606–8612. [Google Scholar] [CrossRef]
- Konings, I.R.; van der Gaast, A.; van der Wijk, L.J.; de Jongh, F.E.; Eskens, F.A.; Sleijfer, S. The addition of pravastatin to chemotherapy in advanced gastric carcinoma: A randomised phase II trial. Eur. J. Cancer 2010, 46, 3200–3204. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, K.H.; Lee, G.K.; Lee, S.-H.; Lim, K.Y.; Joo, J.; Go, Y.J.; Lee, J.S.; Han, J.-Y. Randomized Phase II Study of Afatinib Plus Simvastatin Versus Afatinib Alone in Previously Treated Patients with Advanced Nonadenocarcinomatous Non-small Cell Lung Cancer. Cancer Res. Treat. 2017, 49, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, A.H.; Colditz, G.A.; Rosner, B.; Willett, W.C.; Hankinson, S.E. Serum lipids, lipid-lowering drugs, and the risk of breast cancer. Arch. Intern. Med. 2005, 165, 2264–2271. [Google Scholar] [CrossRef]
- Islam, M.; Yang, H.-C.; Nguyen, P.-A.; Poly, T.N.; Huang, C.-W.; Kekade, S.; Khalfan, A.M.; Debnath, T.; Li, Y.-C.J.; Abdul, S.S. Exploring association between statin use and breast cancer risk: An updated meta-analysis. Arch. Gynecol. Obstet. 2017, 296, 1043–1053. [Google Scholar] [CrossRef]
- McMenamin Murray, L.J.; Hughes, C.M.; Cardwell, C.R. Statin use and breast cancer survival: A nationwide cohort study in Scotland. BMC Cancer 2016, 16, 600. [Google Scholar] [CrossRef]
- Abdullah, M.I.; de Wolf, E.; Jawad, M.J.; Richardson, A. The poor design of clinical trials of statins in oncology may explain their failure—Lessons for drug repurposing. Cancer Treat. Rev. 2018, 69, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Higuchi, T.; Hosomi, K.; Takada, M. Association between statin use and cancer: Data mining of a spontaneous reporting database and a claims database. Int. J. Med. Sci. 2015, 12, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005, 12, 592–602. [Google Scholar] [CrossRef]
- Wolfswinkel, J.F.; Furtmueller, E.; Wilderom, C.P.M. Using grounded theory as a method for rigorously reviewing literature. Eur. J. Inf. Syst. 2013, 22, 45–55. [Google Scholar] [CrossRef]
- Glaser, B.G.; Strauss, A.L. Discovery of Grounded Theory: Strategies for Qualitative Research; Routledge: New York, NY, USA, 2017. [Google Scholar]
- Brown, R.B.; Razzaque, M.S. Dysregulation of phosphate metabolism and conditions associated with phosphate toxicity. BoneKEy Rep. 2015, 4, 705. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. 2018, 150, 398–402. [Google Scholar] [CrossRef]
- Chou, R.; Cantor, A.; Dana, T.; Wagner, J.; Ahmed, A.Y.; Fu, R.; Ferencik, M. Statin Use for the Primary Prevention of Cardiovascular Disease in Adults: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2022, 328, 754–771. [Google Scholar] [CrossRef]
- Byrne, P.; Demasi, M.; Jones, M.; Smith, S.M.; O’Brien, K.K.; DuBroff, R. Evaluating the Association Between Low-Density Lipoprotein Cholesterol Reduction and Relative and Absolute Effects of Statin Treatment: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2022, 182, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Irwig, L.; Irwig, J.; Revena, L.; Sweet, M. Chapter 18, Relative risk, relative and absolute risk reduction, number needed to treat and confidence intervals. In Smart Health Choices: Making Sense of Health Advice; Hammersmith Press: London, UK, 2008. [Google Scholar]
- Brown, R.B. Relative risk reduction: Misinformative measure in clinical trials and COVID-19 vaccine efficacy. Dialogues Health 2022, 1, 100074. [Google Scholar] [CrossRef]
- Schlesselman, J.J. Jerome Cornfield’s Bayesian approach to assessing interim results in clinical trials. J. R. Soc. Med. 2016, 109, 27–35. [Google Scholar] [CrossRef]
- Cornfield, J.; Haenszel, W.; Hammond, E.C.; Lilienfeld, A.M.; Shimkin, M.B.; Wynder, E.L. Smoking and lung cancer: Recent evidence and a discussion of some questions. J. Natl. Cancer Inst. 1959, 22, 173–203. [Google Scholar] [CrossRef] [PubMed]
- Hariton, E.; Locascio, J.J. Randomised controlled trials—The gold standard for effectiveness research: Study design: Randomised controlled trials. Bjog 2018, 125, 1716. [Google Scholar] [CrossRef] [PubMed]
- Principles of Epidemiology in Public Health Practice, Lesson 1: Introduction to Epidemiology. Section 7: Analytic Epidemiology. 2012. Available online: https://www.cdc.gov/csels/dsepd/ss1978/lesson1/section7.html (accessed on 6 December 2021).
- Stadel, B.V.; Colman, E.; Sahlroot, T. Misleading use of risk ratios. Lancet 2005, 365, 1306–1307. [Google Scholar] [CrossRef]
- Doi, S.A.; Furuya-Kanamori, L.; Xu, C.; Lin, L.; Chivese, T.; Thalib, L. Questionable utility of the relative risk in clinical research: A call for change to practice. J. Clin. Epidemiol. 2020, 142, 271–279. [Google Scholar] [CrossRef]
- Fischhoff, B.; Brewer, N.; Downs, J. Communicating Risks and Benefits: An Evidence-Based User’s Guide; Food and Drug Administration (FDA), US Department of Health and Human Services: Silver Spring, MA, USA, 2011. [Google Scholar]
- Mortensen, M.B.; Nordestgaard, B.G. Statin Use in Primary Prevention of Atherosclerotic Cardiovascular Disease According to 5 Major Guidelines for Sensitivity, Specificity, and Number Needed to Treat. JAMA Cardiol. 2019, 4, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Jeeyavudeen, M.S.; Pappachan, J.M.; Arunagirinathan, G. Statin-related Muscle Toxicity: An Evidence-based Review. Touchrev Endocrinol. 2022, 18, 89–95. [Google Scholar] [CrossRef]
- Toth, P.P.; Banach, M. Statins: Then and Now. Methodist. Debakey Cardiovasc. J. 2019, 15, 23–31. [Google Scholar] [CrossRef]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Akyea, R.K.; Kai, J.; Qureshi, N.; Iyen, B.; Weng, S.F. Sub-optimal cholesterol response to initiation of statins and future risk of cardiovascular disease. Heart 2019, 105, 975–981. [Google Scholar] [CrossRef]
- Yebyo, H.G.; Aschmann, H.E.; Kaufmann, M.; Puhan, M.A. Comparative effectiveness and safety of statins as a class and of specific statins for primary prevention of cardiovascular disease: A systematic review, meta-analysis, and network meta-analysis of randomized trials with 94,283 participants. Am. Heart J. 2019, 210, 18–28. [Google Scholar] [CrossRef]
- Carpenter, R.; Waldrop, J.; Carter-Templeton, H. Statistical, practical and clinical significance and Doctor of Nursing Practice projects. Nurse Author Ed. 2021, 31, 50–53. [Google Scholar] [CrossRef]
- Liu, K.; Wilkins, J.T.; Colangelo, L.A.; Lloyd-Jones, D.M. Does Lowering Low-Density Lipoprotein Cholesterol With Statin Restore Low Risk in Middle-Aged Adults? Analysis of the Observational MESA Study. J. Am. Heart Assoc. 2021, 10, e019695. [Google Scholar] [CrossRef]
- Fernandez-Friera, L.; Fuster, V.; López-Melgar, B.; Oliva, B.; García-Ruiz, J.M.; Mendiguren, J.; Bueno, H.; Pocock, S.; Ibanez, B.; Fernández-Ortiz, A.; et al. Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. J. Am. Coll. Cardiol. 2017, 70, 2979–2991. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.L.; Lambert, G.; Mach, F.; et al. Frequent questions and responses on the 2022 lipoprotein(a) consensus statement of the European Atherosclerosis Society. Atherosclerosis 2023, 374, 107–120. [Google Scholar] [CrossRef]
- Brown, R.B. Phospholipid packing defects and oxysterols in atherosclerosis: Dietary prevention and the French paradox. Biochimie 2019, 167, 145–151. [Google Scholar] [CrossRef]
- Virchow, R. As Based upon Physiological and Pathological Histology. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- Ross, R.; Harker, L. Hyperlipidemia and atherosclerosis. Science 1976, 193, 1094–1100. [Google Scholar] [CrossRef]
- Gerrity, R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am. J. Pathol. 1981, 103, 181–190. [Google Scholar]
- Tucker, W.D.; Arora, Y.; Mahajan, K. Anatomy, Blood Vessels. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Falk, E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br. Heart J. 1983, 50, 127–134. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.-K. Reassessing the Mechanisms of Acute Coronary Syndromes: The “vulnerable plaque” and superficial erosion. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Glagov, S.; Weisenberg, E.; Zarins, C.K.; Stankunavicius, R.; Kolettis, G.J. Compensatory enlargement of human atherosclerotic coronary arteries. N. Engl. J. Med. 1987, 316, 1371–1375. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The role of lipids and lipoproteins in atherosclerosis. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Keys, A. Coronary heart disease in seven countries. Nutrition 1997, 13, 249–253; discussion 249, 253. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Nair, P. Brown and Goldstein: The cholesterol chronicles. Proc. Natl. Acad. Sci. USA 2013, 110, 14829–14832. [Google Scholar] [CrossRef]
- Kulig, W.; Cwiklik, L.; Jurkiewicz, P.; Rog, T.; Vattulainen, I. Cholesterol oxidation products and their biological importance. Chem. Phys. Lipids 2016, 199, 144–160. [Google Scholar] [CrossRef]
- Olkkonen, V.M.; Hynynen, R. Interactions of oxysterols with membranes and proteins. Mol. Asp. Med. 2009, 30, 123–133. [Google Scholar] [CrossRef]
- Pinot, M.; Vanni, S.; Ambroggio, E.; Guet, D.; Goud, B.; Manneville, J.-B. Feedback between membrane tension, lipid shape and curvature in the formation of packing defects. bioRxiv 2018. [Google Scholar] [CrossRef]
- Bigay, J.; Antonny, B. Curvature, Lipid Packing, and Electrostatics of Membrane Organelles: Defining Cellular Territories in Determining Specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef]
- Zhang, X.; Sessa, W.C.; Fernández-Hernando, C. Endothelial transcytosis of lipoproteins in atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 130. [Google Scholar] [CrossRef]
- Pordal, A.-H.; Hajmiresmail, S.J.; Assadpoor-Piranfar, M.; Hedayati, M.; Ajami, M. Plasma oxysterol level in patients with coronary artery stenosis and its changes in response to the treatment with atorvastatin. Med. J. Islam. Repub. Iran. 2015, 29, 192. [Google Scholar]
- Song, J.; Wang, D.; Chen, H.; Huang, X.; Zhong, Y.; Jiang, N.; Chen, C.; Xia, M. Association of plasma 7-ketocholesterol with cardiovascular outcomes and total mortality in patients with coronary artery disease. Circ. Res. 2017, 120, 1622–1631. [Google Scholar] [CrossRef]
- Min, J.-S.; Lee, S.-O.; Khan, M.I.; Yim, D.G.; Seol, K.-H.; Lee, M.; Jo, C. Monitoring the formation of cholesterol oxidation products in model systems using response surface methodology. Lipids Health Dis. 2015, 14, 77. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Xiao, F.; Jie, F.; Zhang, Q.; Liu, Y.; Xiao, H.; Lu, B. Dietary cholesterol oxidation products: Perspectives linking food processing and storage with health implications. Compr. Rev. Food Sci. Food Saf. 2022, 21, 738–779. [Google Scholar] [CrossRef] [PubMed]
- Kahleova, H.; Levin, S.; Barnard, N. Cardio-metabolic benefits of plant-based diets. Nutrients 2017, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Ellis, F.; Sanders, T. Angina and vegan diet. Am. Heart J. 1977, 93, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Ornish, D.; Brown, S.E.; Billings, J.H.; Scherwitz, L.W.; Armstrong, W.T.; Ports, T.A.; McLanahan, S.M.; Kirkeeide, R.L.; Gould, K.L.; Brand, R.J. Can lifestyle changes reverse coronary heart disease?: The Lifestyle Heart Trial. Lancet 1990, 336, 129–133. [Google Scholar] [CrossRef]
- Bonovas, S.; Sitaras, N.M. Does pravastatin promote cancer in elderly patients? A meta-analysis. Cmaj 2007, 176, 649–654. [Google Scholar] [CrossRef]
- Tonkin, A.M.; Forbes, A.; Haas, S.J. The evidence on trial: Cholesterol lowering and cancer. Heart Asia 2009, 1, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.B.; Hulley, S.B. Carcinogenicity of lipid-lowering drugs. JAMA 1996, 275, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.O.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Rossebø, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Bärwolf, C.; Holme, I.; Kesäniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef]
- Kaye, J.A.; Jick, H. Statin use and cancer risk in the General Practice Research Database. Br. J. Cancer 2004, 90, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Agalliu, I.; Salinas, C.A.; Hansten, P.D.; Ostrander, E.A.; Stanford, J.L. Statin use and risk of prostate cancer: Results from a population-based epidemiologic study. Am. J. Epidemiol. 2008, 168, 250–260. [Google Scholar] [CrossRef]
- Vinogradova, Y.; Coupland, C.; Hippisley-Cox, J. Exposure to statins and risk of common cancers: A series of nested case-control studies. BMC Cancer 2011, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Ho, S.C.; Chiu, H.F.; Yang, C.Y. Statins increase the risk of prostate cancer: A population-based case-control study. Prostate 2011, 71, 1818–1824. [Google Scholar] [CrossRef]
- McDougall, J.A.; Malone, K.E.; Daling, J.R.; Cushing-Haugen, K.L.; Porter, P.L.; Li, C.I. Long-term statin use and risk of ductal and lobular breast cancer among women 55 to 74 years of age. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1529–1537. [Google Scholar] [CrossRef]
- Desai, P.; Wallace, R.; Anderson, M.L.; Howard, B.V.; Ray, R.M.; Wu, C.; Safford, M.; Martin, L.W.; Rohan, T.; Manson, J.E.; et al. An analysis of the association between statin use and risk of endometrial and ovarian cancers in the Women’s Health Initiative. Gynecol. Oncol. 2018, 148, 540–546. [Google Scholar] [CrossRef]
- Mamtani, R.; Lewis, J.D.; Scott, F.I.; Ahmad, T.; Goldberg, D.S.; Datta, J.; Yang, Y.-X.; Boursi, B. Disentangling the Association between Statins, Cholesterol, and Colorectal Cancer: A Nested Case-Control Study. PLoS Med. 2016, 13, e1002007. [Google Scholar] [CrossRef]
- Aronson, J.; Bankhead, C.; Mahtani, K.; Nunan, D. Confounding by Indication. Catalogue of Biases. 2018. Available online: https://catalogofbias.org/biases/confounding-by-indication/#:~:text=A%20distortion%20that%20modifies%20an,true%20cause%20of%20the%20outcome (accessed on 16 September 2024).
- Brown, R.B. Phosphate and oxysterols may mediate an inverse relationship between atherosclerosis and cancer. Eur. Med. J.-Oncol. 2020, 8, 114–121. [Google Scholar] [CrossRef]
- Elkeles, A. Cancer and atherosclerosis. Br. J. Cancer 1956, 10, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Benito-León, J.; Aleja, J.G.; Martínez-Salio, A.; Louis, E.D.; Lichtman, J.H.; Bermejo-Pareja, F. Symptomatic Atherosclerotic Disease and Decreased Risk of Cancer-Specific Mortality: A Prospective, Population-Based Study (NEDICES). Medicine 2015, 94, e1287. [Google Scholar] [CrossRef] [PubMed]
- Jirasek, A.; Knief, J.; Deng, M.; Reddemann, K.; Thorns, C. Evaluation of general and coronary atherosclerosis and malignant disease demonstrates inverse correlations for specific cancer types as well as cancer in general. Pathol. Res. Pract. 2016, 212, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cima, M.J.; Milner, D.A., Jr. If It’s Not One Thing, It’s Another: An Inverse Relationship of Malignancy and Atherosclerotic Disease. PLoS ONE 2015, 10, e0126855. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Phosphate toxicity and tumorigenesis. Biochim. Biophys. Acta (BBA)–Rev. Cancer 2018, 1869, 303–309. [Google Scholar] [CrossRef]
- Lacerda-Abreu, M.A.; Meyer-Fernandes, J.R. Elevated extracellular inorganic phosphate inhibits ecto-phosphatase activity in breast cancer cells: Regulation by hydrogen peroxide. Cell Biol. Int. 2024, 48, 162–173. [Google Scholar] [CrossRef]
- D’Arcangelo, M.; Brustugun, O.T.; Xiao, Y.; Choi, Y.; Behrens, C.; Solis, L.M.; Wang, Y.; Firestein, R.; Boyle, T.A.; Lund-Iversen, M.; et al. 194P prevalence and prognostic significance of sodium-dependent phosphate transporter 2B (NAPI2B) protein expression in non-small cell lung cancer (NSCLC). Ann. Oncol. 2014, 25 (Suppl. 4), iv66. [Google Scholar] [CrossRef]
- Levan, K.; Mehryar, M.; Mateoiu, C.; Albertsson, P.; Bäck, T.; Sundfeldt, K. Immunohistochemical evaluation of epithelial ovarian carcinomas identifies three different expression patterns of the MX35 antigen, NaPi2b. BMC Cancer 2017, 17, 303. [Google Scholar] [CrossRef] [PubMed]
- Elser, J.J.; Kyle, M.M.; Smith, M.S.; Nagy, J.D. Biological stoichiometry in human cancer. PLoS ONE 2007, 2, e1028. [Google Scholar] [CrossRef] [PubMed]
- Bobko, A.A.; Eubank, T.D.; Driesschaert, B.; Dhimitruka, I.; Evans, J.; Mohammad, R.; Tchekneva, E.E.; Dikov, M.M.; Khramtsov, V.V. Interstitial inorganic phosphate as a tumor microenvironment marker for tumor progression. Sci. Rep. 2017, 7, 41233. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.N.; Griffin, A.C. Phosphorus incorporation into nucleic acids and proteins of liver nuclei of normal and azo dye-fed rats. Cancer Res. 1955, 15, 456–461. [Google Scholar] [PubMed]
- Lin, Y.; McKinnon, K.E.; Ha, S.W.; Beck, G.R., Jr. Inorganic phosphate induces cancer cell mediated angiogenesis dependent on forkhead box protein C2 (FOXC2) regulated osteopontin expression. Mol. Carcinog. 2015, 54, 926–934. [Google Scholar] [CrossRef]
- Jin, H.; Xu, C.-X.; Lim, H.-T.; Park, S.-J.; Shin, J.-Y.; Chung, Y.-S.; Park, S.-C.; Chang, S.-H.; Youn, H.-J.; Lee, K.-H.; et al. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 59–68. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Camalier, C.E.; Young, M.R.; Bobe, G.; Perella, C.M.; Colburn, N.H.; Beck, G.R. Elevated phosphate activates N-ras and promotes cell transformation and skin tumorigenesis. Cancer Prev. Res. 2010, 3, 359–370. [Google Scholar] [CrossRef]
- Wilson, K.M.; Shui, I.M.; Mucci, L.A.; Giovannucci, E. Calcium and phosphorus intake and prostate cancer risk: A 24-y follow-up study. Am. J. Clin. Nutr. 2015, 101, 173–183. [Google Scholar] [CrossRef]
- McClure, S.T.; Chang, A.R.; Selvin, E.; Rebholz, C.M.; Appel, L.J. Dietary Sources of Phosphorus among Adults in the United States: Results from NHANES 2001-2014. Nutrients 2017, 9, 95. [Google Scholar] [CrossRef]
- Michaëlsson, K.; Wolk, A.; Langenskiöld, S.; Basu, S.; Lemming, E.W.; Melhus, H.; Byberg, L. Milk intake and risk of mortality and fractures in women and men: Cohort studies. BMJ Br. Med. J. 2014, 349, g6015. [Google Scholar] [CrossRef] [PubMed]
- Papaloucas, C.; Papaloucas; Kouloulias, V.; Neanidis, K.; Pistevou-Gompaki, K.; Kouvaris, J.; Zygogianni, A.; Mystakidou, K.; Papaloucas, A. Measurement of blood phosphorus: A quick and inexpensive method for detection of the existence of cancer in the body. Too good to be true, or forgotten knowledge of the past? Med. Hypotheses 2014, 82, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Wulaningsih, W.; Michaelsson, K.; Garmo, H.; Hammar, N.; Jungner, I.; Walldius, G.; Holmberg, L.; Van Hemelrijck, M. Inorganic phosphate and the risk of cancer in the Swedish AMORIS study. BMC Cancer 2013, 13, 257. [Google Scholar] [CrossRef]
- Kouloulias, V.; Tolia, M.; Tsoukalas, N.; Papaloucas, C.; Pistevou-Gombaki, K.; Zygogianni, A.; Mystakidou, K.; Kouvaris, J.; Papaloucas, M.; Psyrri, A.; et al. Is there any potential clinical impact of serum phosphorus and magnesium in patients with lung cancer at first diagnosis? A multi-institutional study. Asian Pac. J. Cancer Prev. 2015, 16, 77–81. [Google Scholar] [CrossRef]
- Ye, Z.; Palazzo, J.P.; Lin, L.; Lai, Y.; Guiles, F.; E Myers, R.; Han, J.; Xing, J.; Yang, H. Postoperative hyperphosphatemia significantly associates with adverse survival in colorectal cancer patients. J. Gastroenterol. Hepatol. 2013, 28, 1469–1475. [Google Scholar] [CrossRef]
- Kuang, Y.; Nagy, J.D.; Elser, J.J. Biological stoichiometry of tumor dynamics: Mathematical models and analysis. Discret. Contin. Dyn. Syst. Ser. B 2004, 4, 221–240. [Google Scholar]
- Boyineni, J.; Sredni, S.T.; Margaryan, N.V.; Demirkhanyan, L.; Tye, M.; Johnson, R.; Gonzalez-Nilo, F.; Hendrix, M.J.; Pavlov, E.; Soares, M.B.; et al. Inorganic polyphosphate as an energy source in tumorigenesis. Oncotarget 2020, 11, 4613–4624. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef]
- Lau, W.M.; Doucet, M.; Stadel, R.; Huang, D.; Weber, K.L.; Kominsky, S.L. Enpp1: A Potential Facilitator of Breast Cancer Bone Metastasis. PLoS ONE 2013, 8, e66752. [Google Scholar] [CrossRef]
- Brown, R.B. Spontaneous Tumor Regression and Reversion: Insights and Associations with Reduced Dietary Phosphate. Cancers 2024, 16, 2126. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, C.; Piccoli, G.B.; Cupisti, A. The “phosphorus pyramid”: A visual tool for dietary phosphate management in dialysis and CKD patients. BMC Nephrol. 2015, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Food Data Central 2023. Available online: https://fdc.nal.usda.gov/ (accessed on 12 July 2023).
- Wanscher, O.; Clemmesen, J.; Nielsen, A. Negative Correlation Between Atherosclerosis and Carcinoma. Br. J. Cancer 1951, 5, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Juhl, S. Cancer and atherosclerosis. 2. Applicability of postmortem statistics in the study of the negative correlation. Acta Pathol. Microbiol. Scand. 1957, 41, 99–104. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Luckoor, P.; Salehi, M.; Kunadu, A. Exceptionally High Creatine Kinase (CK) Levels in Multicausal and Complicated Rhabdomyolysis: A Case Report. Am. J. Case Rep. 2017, 18, 746–749. [Google Scholar] [CrossRef]
- Cabral, B.M.I.; Edding, S.N.; Portocarrero, J.P.; Lerma, E.V. Rhabdomyolysis. Dis. Mon. 2020, 66, 101015. [Google Scholar] [CrossRef]
- Stahl, K.; Rastelli, E.; Schoser, B. A systematic review on the definition of rhabdomyolysis. J. Neurol. 2020, 267, 877–882. [Google Scholar] [CrossRef]
- Gupta, A.; Moore, J.A. Tumor Lysis Syndrome. JAMA Oncol. 2018, 4, 895. [Google Scholar] [CrossRef]
- Chavez, L.O.; Leon, M.; Einav, S.; Varon, J. Beyond muscle destruction: A systematic review of rhabdomyolysis for clinical practice. Crit. Care 2016, 20, 135. [Google Scholar] [CrossRef]
- Hird, A.E.; Magee, D.E.; Matta, R.; Saskin, R.; Dvorani, E.; Kulkarni, G.S.; Kodama, R.; Herschorn, S.; Narod, S.A.; Nam, R.K. Assessment of Secondary Sarcomas Among Patients With Cancer of the Abdomen or Pelvis Who Received Combinations of Surgery, Radiation, and Chemotherapy vs Surgery Alone. JAMA Netw. Open 2020, 3, e2013929. [Google Scholar] [CrossRef] [PubMed]
- Safitri, N.; Alaina, M.F.; Pitaloka, D.A.E.; Abdulah, R. A Narrative Review of Statin-Induced Rhabdomyolysis: Molecular Mechanism, Risk Factors, and Management. Drug Healthc. Patient Saf. 2021, 13, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Westwood, F.R.; Bigley, A.; Randall, K.; Marsden, A.M.; Scott, R.C. Statin-induced muscle necrosis in the rat: Distribution, development, and fibre selectivity. Toxicol. Pathol. 2005, 33, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.A.; Helmstetter, J.A.; Kaye, A.M.; Kaye, A.D. Rhabdomyolysis: Pathogenesis, diagnosis, and treatment. Ochsner J. 2015, 15, 58–69. [Google Scholar]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1046–e1081. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Ezad, S.; Cheema, H.; Collins, N. Statin-induced rhabdomyolysis: A complication of a commonly overlooked drug interaction. Oxf. Med. Case Rep. 2018, 2018, omx104. [Google Scholar] [CrossRef]
- Omar, M.A.; Wilson, J.P. FDA Adverse Event Reports on Statin-Associated Rhabdomyolysis. Ann. Pharmacother. 2002, 36, 288–295. [Google Scholar] [CrossRef]
- Bakhai, A.; Rigney, U.; Hollis, S.; Emmas, C. Co-administration of statins with cytochrome P450 3A4 inhibitors in a UK primary care population. Pharmacoepidemiol. Drug Saf. 2012, 21, 485–493. [Google Scholar] [CrossRef]
- Montastruc, J.-L. Rhabdomyolysis and statins: A pharmacovigilance comparative study between statins. Br. J. Clin. Pharmacol. 2023, 89, 2636–2638. [Google Scholar] [CrossRef]
- Sattar, N. Statins and diabetes: What are the connections? Best. Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101749. [Google Scholar] [CrossRef]
- Brown, R.B. Diabetes, diabetic complications, and phosphate toxicity: A scoping review. Curr. Diabetes Rev. 2020, 16, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.A.; Campana-Salort, E.; Begu-LeCorroller, A.; Baccou, M.; Valéro, R.; Vialettes, B. Non-traumatic rhabdomyolysis and diabetes. Diabetes Metab. 2011, 37, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sterling, N.W.; Kong, L.; Lewis, M.M.; Mailman, R.B.; Chen, H.; Leslie, D.; Huang, X. Statins may facilitate Parkinson’s disease: Insight gained from a large, national claims database. Mov. Disord. 2017, 32, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B. Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity. Int. J. Mol. Sci. 2022, 23, 8060. [Google Scholar] [CrossRef]
- Langsjoen, P.H.; Langsjoen, J.O.; Langsjoen, A.M.; Rosenfeldt, F. Statin-Associated Cardiomyopathy Responds to Statin Withdrawal and Administration of Coenzyme Q(10). Perm. J. 2019, 23, 8060. [Google Scholar] [CrossRef]
- Patail, H.; Kothari, A.; Nadig, V.; Kunkes, J. Statin-Induced Myositis with Concomitant Myocarditis. Cureus 2022, 14, e31871. [Google Scholar] [CrossRef]
- Hamel, Y.; Mamoune, A.; Mauvais, F.X.; Habarou, F.; Lallement, L.; Romero, N.B.; Ottolenghi, C.; de Lonlay, P. Acute rhabdomyolysis and inflammation. J. Inherit. Metab. Dis. 2015, 38, 621–628. [Google Scholar] [CrossRef]
- Subashri, M.; Sujit, S.; Thirumalvalavan, K.; Poongodi, A.; Srinivasaprasad, N.D.; Edwin Fernando, M. Rhabdomyolysis-associated Acute Kidney Injury. Indian J. Nephrol. 2023, 33, 114–118. [Google Scholar] [CrossRef]
- Acharya, T.; Huang, J.; Tringali, S.; Frei, C.R.; Mortensen, E.M.; Mansi, I.A. Statin Use and the Risk of Kidney Disease with Long-Term Follow-Up (8.4-Year Study). Am. J. Cardiol. 2016, 117, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Slatopolsky, E. Phosphate Toxicity in CKD: The Killer among Us. Clin. J. Am. Soc. Nephrol. 2016, 11, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Morse, L.R.; Coker, J.; Battaglino, R.A. Statins and bone health: A mini review. Actual Osteol 2018, 14, 31–35. [Google Scholar] [PubMed]
- Shi, R.; Mei, Z.; Zhang, Z.; Zhu, Z. Effects of Statins on Relative Risk of Fractures for Older Adults: An Updated Systematic Review With Meta-Analysis. J. Am. Med. Dir. Assoc. 2019, 20, 1566–1578.e3. [Google Scholar] [CrossRef]
- Goyal, R.; Jialal, I. Hyperphosphatemia; StatPearls Publishing: Tampa, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK551586/ (accessed on 1 April 2024).
- Jiang, Y.; Wang, Y.; Zhao, J.; Marchal, G.; Wang, Y.; Shen, Y.; Xing, S.; Zhang, X.; Baert, A.L. Metastatic calcification within bone. The main cause of osteosclerosis in hypervitaminosis D3. Radiologic-pathologic correlation. Invest. Radiol. 1990, 25, 1188–1196. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Zhou, F.; Piao, Z.; Hao, J. Effects of Statins on Bone Mineral Density and Fracture Risk: A PRISMA-compliant Systematic Review and Meta-Analysis. Medicine 2016, 95, e3042. [Google Scholar] [CrossRef]
- Hardcastle, S.A.; Gregson, C.L.; Deere, K.C.; Davey Smith, G.; Dieppe, P.; Tobias, J.H. High bone mass is associated with an increased prevalence of joint replacement: A case-control study. Rheumatology 2013, 52, 1042–1051. [Google Scholar] [CrossRef]
- Daba, K.T.; Weldemichael, D.K.; Mulugeta, G.A. Bilateral hypocalcemic cataract after total thyroidectomy in a young woman: Case report. BMC Ophthalmol. 2019, 19, 233. [Google Scholar] [CrossRef] [PubMed]
- Ghouse, J.; Ahlberg, G.; Skov, A.G.; Bundgaard, H.; Olesen, M.S. Association of Common and Rare Genetic Variation in the 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Gene and Cataract Risk. J. Am. Heart Assoc. 2022, 11, e025361. [Google Scholar] [CrossRef]
- Henein, M.; Granåsen, G.; Wiklund, U.; Schmermund, A.; Guerci, A.; Erbel, R.; Raggi, P. High dose and long-term statin therapy accelerate coronary artery calcification. Int. J. Cardiol. 2015, 184, 581–586. [Google Scholar] [CrossRef]
- Dykun, I.; Lehmann, N.; Kälsch, H.; Möhlenkamp, S.; Moebus, S.; Budde, T.; Seibel, R.; Grönemeyer, D.; Jöckel, K.H.; Erbel, R.; et al. Statin Medication Enhances Progression of Coronary Artery Calcification: The Heinz Nixdorf Recall Study. J. Am. Coll. Cardiol. 2016, 68, 2123–2125. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B. Vitamin D, cancer, and dysregulated phosphate metabolism. Endocrine 2019, 65, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Malatesta, K.; Norris, K. Vitamin D and chronic kidney disease. Ethn. Dis. 2009, 19 (Suppl. 5), S5. [Google Scholar] [PubMed]
- Pennisi, M.; Di Bartolo, G.; Malaguarnera, G.; Bella, R.; Lanza, G.; Malaguarnera, M. Vitamin D Serum Levels in Patients with Statin-Induced Musculoskeletal Pain. Dis. Markers 2019, 2019, 3549402. [Google Scholar] [CrossRef]
- Mazidi, M.; Rezaie, P.; Vatanparast, H.; Kengne, A.P. Effect of statins on serum vitamin D concentrations: A systematic review and meta-analysis. Eur. J. Clin. Investig. 2017, 47, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Peyrel, P.; Mauriège, P.; Frenette, J.; Laflamme, N.; Greffard, K.; Dufresne, S.S.; Huth, C.; Bergeron, J.; Joanisse, D.R. No benefit of vitamin D supplementation on muscle function and health-related quality of life in primary cardiovascular prevention patients with statin-associated muscle symptoms: A randomized controlled trial. J. Clin. Lipidology. 2023, 18, e269–e284. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, M.A.; Gonzalez, P.E.; Manson, J.E.; Buring, J.E.; Lee, I.-M.; Cook, N.R.; Mora, S.; Bubes, V.; Stone, N.J. Statin-Associated Muscle Symptoms Among New Statin Users Randomly Assigned to Vitamin D or Placebo. JAMA Cardiol. 2023, 8, 74–80. [Google Scholar] [CrossRef]
- Brown, R.B. Dysregulated Phosphate Metabolism, Periodontal Disease, and Cancer: Possible Global Health Implications. Dent. J. 2019, 7, 18. [Google Scholar] [CrossRef]
- Petit, C.; Batool, F.; Bugueno, I.M.; Schwinté, P.; Benkirane-Jessel, N.; Huck, O. Contribution of Statins towards Periodontal Treatment: A Review. Mediat. Inflamm. 2019, 2019, 6367402. [Google Scholar] [CrossRef]
- Kwon, M.J.; Byun, S.-H.; Kim, J.-H.; Kim, J.H.; Kim, S.H.; Kim, N.Y.; Park, H.-R.; Choi, H.G. Longitudinal follow-up study of the association between statin use and chronic periodontitis using national health screening cohort of Korean population. Sci. Rep. 2022, 12, 5504. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, C.; Ma, X.; Wu, Q.; Zhou, F.; Liu, X. The association between statin use and osteoarthritis-related outcomes: An updated systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 1003370. [Google Scholar] [CrossRef]
- Julovi, S.M.; Dao, A.; Trinh, K.; O’donohue, A.K.; Shu, C.; Smith, S.; Shingde, M.; Schindeler, A.; Rogers, N.M.; Little, C.B. Disease-modifying interactions between chronic kidney disease and osteoarthritis: A new comorbid mouse model. RMD Open 2023, 9, e003109. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.N.; Gillman, B.A.; Kiely, M.; Palmer, B.; Shiely, F.; Kearney, P.M.; Earlie, J.; Bowles, M.B.; Keohane, F.M.; Connolly, P.P.; et al. Pilot Randomized Controlled Trial of a Standard Versus a Modified Low-Phosphorus Diet in Hemodialysis Patients. Kidney Int. Rep. 2020, 5, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B.; Bigelow, P. Can a Low-Phosphate Diet for Chronic Kidney Disease Treat Cancer? An Interdisciplinary Literature Review. Medicines 2024, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.V.; Finisguerra, V.; Van den Eynde, B.J.; Bercovici, N.; Trautmann, A. Preclinical murine tumor models: A structural and functional perspective. Elife 2020, 9, e50740. [Google Scholar] [CrossRef]
- Gisterå, A.; Ketelhuth, D.F.J.; Malin, S.G.; Hansson, G.K. Animal Models of Atherosclerosis–Supportive Notes and Tricks of the Trade. Circ. Res. 2022, 130, 1869–1887. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Risk | The percentage of an event/outcome/endpoint in a clinical trial that occurs in each of two groups, the experimental group receiving a treatment/intervention and the control group receiving a placebo. |
| Relative Risk | The ratio of the experimental group risk relative to (divided by) the control group risk (the baseline risk). |
| Relative Risk Reduction | The relative risk subtracted from the null value of 1.00. The null value has equal risks in both groups. |
| Absolute Risk Reduction | The experimental group risk subtracted from the control group risk, indicating the size of the risk difference. |
| Relative Risk Reduction (alternative calculation) | The absolute risk reduction is divided by the baseline risk. |
| Number Needed to Treat | The null value (1.00) divided by the absolute risk reduction (i.e., the reciprocal of the absolute risk reduction) which indicates the number of treated patients needed to reduce one event. |
| Reviewed Evidence | Knowledge Synthesis |
|---|---|
| Statin therapy is associated with cancer protection [6,7], plausibly due to confounding by indication for statin therapy [89]. | Confounding by indication for statin therapy is potentially caused by an inverse relationship between cancer and atherosclerosis [91]. |
| Statin therapy is associated with increased cancer risk [79,87,88]. Rhabdomyolysis is also an adverse effect of statin therapy which breaks down skeletal muscle [126]. | Phosphate released into serum from rhabdomyolysis increases the risk of phosphate toxicity and subsequent increased risk of tumorigenesis [100,101]. |
| High relative risk reductions of cardiovascular disease are reported in clinical trials of statin therapies [28,42,45]. But low absolute risk reductions and low clinical effect sizes of statin therapy are often unreported in clinical trials [29,30]. | The U.S. FDA and HHS advise reporting both relative and absolute risk reductions in clinical trials [39], which suggests that the putative benefits of statin therapy may not outweigh adverse effects. |
| Statin use is strongly associated with comorbidities: e.g., diabetes [141], Parkinson’s disease [144], cardiomyopathy [146], and kidney injury [150]. | Phosphate toxicity and related rhabdomyolysis potentially mediate the association of statins with comorbidities: e.g., diabetes [142], Parkinson’s disease [144], inflammation [148], and CKD [151]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, R.B. Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity. J. Cardiovasc. Dev. Dis. 2024, 11, 296. https://doi.org/10.3390/jcdd11090296
Brown RB. Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity. Journal of Cardiovascular Development and Disease. 2024; 11(9):296. https://doi.org/10.3390/jcdd11090296
Chicago/Turabian StyleBrown, Ronald B. 2024. "Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity" Journal of Cardiovascular Development and Disease 11, no. 9: 296. https://doi.org/10.3390/jcdd11090296
APA StyleBrown, R. B. (2024). Statins in the Cause and Prevention of Cancer: Confounding by Indication and Mediation by Rhabdomyolysis and Phosphate Toxicity. Journal of Cardiovascular Development and Disease, 11(9), 296. https://doi.org/10.3390/jcdd11090296