


Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review

 , , and
, , and
Abstract
:1. Introduction
2. Epidemiology
3. Genetics
3.1. Desmosomal Mutations
3.2. Non-Desmosomal Mutations
3.3. Effect Modifiers
4. Pathophysiology
4.1. Arrhythmogenesis
4.2. Adipogenesis
4.3. Cardiomyocyte Mechanical Injury, Apoptosis, and Necrosis
4.4. Myocardial Fibrosis and Inflammation
5. Diagnosis
5.1. Clinical Presentation
5.1.1. Asymptomatic
5.1.2. Arrhythmias
5.1.3. Sudden Cardiac Death
5.1.4. Syncope
5.1.5. Ventricular Dysfunction
5.1.6. Extracardiac Findings
5.2. Diagnostic Tools: Genetic Testing, Histopathology, EKG, Echo, cMRI, EP Study
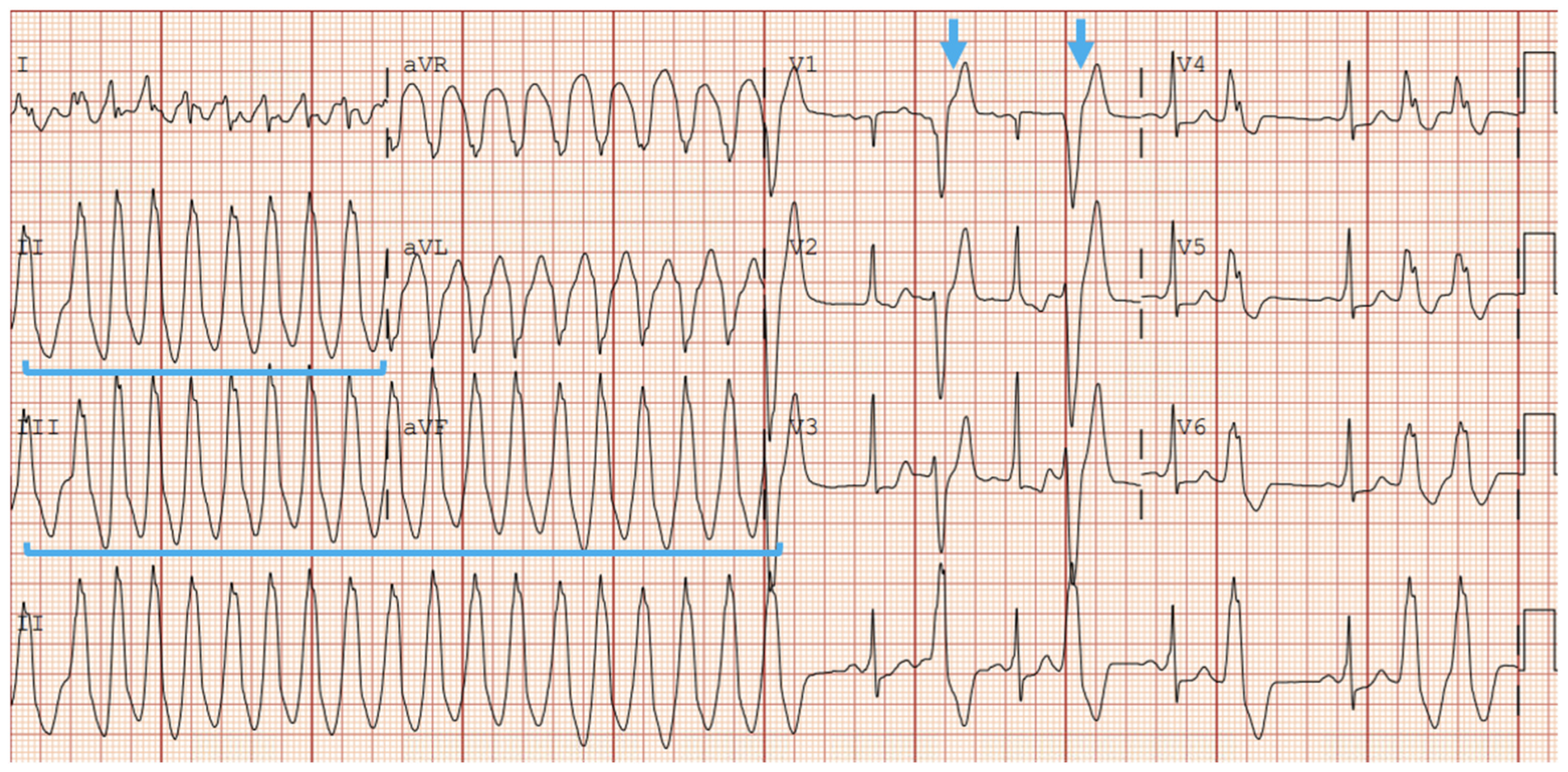
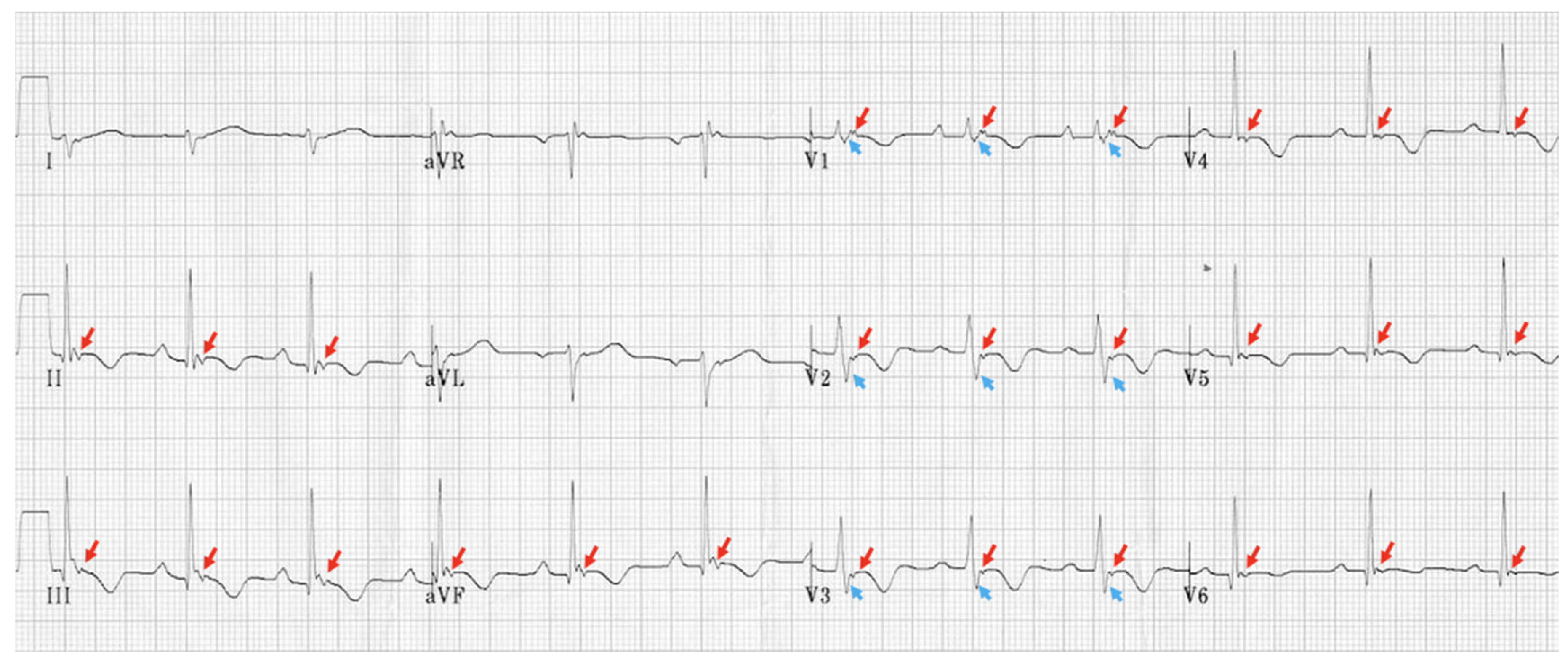
5.2.1. EKG and Holter Monitoring
5.2.2. Echocardiogram
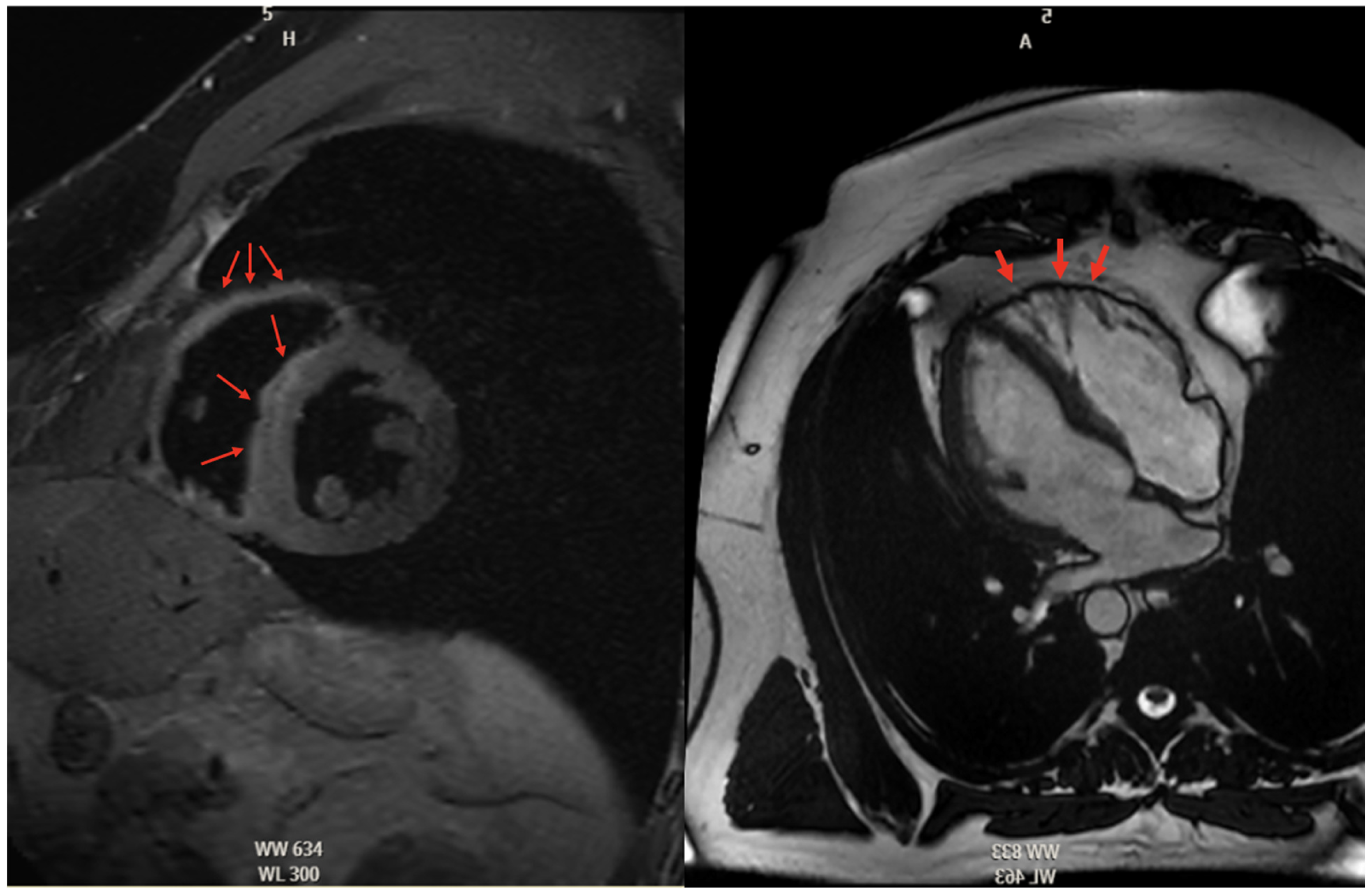
5.2.3. Cardiac Magnetic Resonance High Risk
5.2.4. Histopathologic Findings in ARVC
5.2.5. Role of Electrophysiologic Studies
5.2.6. Multi-Modal Imaging
5.2.7. Genetic Testing in ARVC
5.3. Diagnostic Criteria
6. Treatment: Anti-Arrhythmics, ICD Placement, Ablation, Standard of Care
6.1. Conservative Treatment
6.2. Pharmacologic Therapy
6.3. Implantable Cardioverter-Defibrillator
6.4. Ablation
6.5. Cardiac Transplantation
7. Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Asatryan, B.; Siripanthong, B.; Munroe, P.B.; Tiku-Owens, A.; Lopes, L.R.; Khanji, M.Y.; Protonotarios, A.; Santangeli, P.; Muser, D.; et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6615. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Wang, W.; James, C.A.; Calkins, H. Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace 2019, 21, 9–21. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- Coonar, A.S.; Protonotarios, N.; Tsatsopoulou, A.; Needham, E.W.; Houlston, R.S.; Cliff, S.; Otter, M.I.; Murday, V.A.; Mattu, R.K.; McKenna, W.J. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998, 97, 2049–2058. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.D.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; te Riele, A.S.J.M.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.J.M.; James, C.A.; Philips, B.; Rastegar, N.; Bhonsale, A.; Groeneweg, J.A.; Murray, B.; Tichnell, C.; Judge, D.P.; Van Der Heijden, J.F.; et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: The triangle of dysplasia displaced. J. Cardiovasc. Electrophysiol. 2013, 24, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Judge, D.P. Epidemiology of the inherited cardiomyopathies. Nat. Rev. Cardiol. 2021, 18, 22–36. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Quarta, G.; Muir, A.; Pantazis, A.; Syrris, P.; Gehmlich, K.; Garcia-Pavia, P.; Ward, D.; Sen-Chowdhry, S.; Elliott, P.M.; McKenna, W.J. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: Impact of genetics and revised task force criteria. Circulation 2011, 123, 2701–2709. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.M.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.A.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.A.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- Dominguez, F.; Zorio, E.; Jimenez-Jaimez, J.; Salguero-Bodes, R.; Zwart, R.; Gonzalez-Lopez, E.; Molina, P.; Bermúdez-Jiménez, F.; Delgado, J.F.; Braza-Boïls, A.; et al. Clinical characteristics and determinants of the phenotype in TMEM43 arrhythmogenic right ventricular cardiomyopathy type 5. Heart Rhythm 2020, 17, 945–954. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.H.; Wiesfeld, A.C.P.; Jongbloed, J.D.H.; van den Wijngaard, A.; Kuks, J.B.M.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.P.J.; Asimaki, A.; van Rijen, H.V.M.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.G.; et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2013, 10, 412–419. [Google Scholar] [CrossRef]
- Chen, X.; Chen, L.; Chen, Z.; Chen, X.; Song, J. Remodelling of myocardial intercalated disc protein connexin 43 causes increased susceptibility to malignant arrhythmias in ARVC/D patients. Forensic Sci. Int. 2017, 275, 14–22. [Google Scholar] [CrossRef]
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.J. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. J. Cell Biol. 2014, 206, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patiño, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Lorenzon, A.; Calore, M.; Poloni, G.; De Windt, L.J.; Braghetta, P.; Rampazzo, A. Wnt/β-catenin pathway in arrhythmogenic cardiomyopathy. Oncotarget 2017, 8, 60640–60655. [Google Scholar] [CrossRef]
- Ellawindy, A.; Satoh, K.; Sunamura, S.; Kikuchi, N.; Suzuki, K.; Minami, T.; Ikeda, S.; Tanaka, S.; Shimizu, T.; Enkhjargal, B.; et al. Rho-Kinase Inhibition During Early Cardiac Development Causes Arrhythmogenic Right Ventricular Cardiomyopathy in Mice. Arter. Thromb. Vasc. Biol. 2015, 35, 2172–2184. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Zuo, Y.; Farmer, S.R. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol 2006, 26, 5827–5837. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, S.; Dong, T.; Yang, J.; Xie, Y.; Wu, Y.; Kang, K.; Hu, S.; Gou, D.; Wei, Y. Profiling of differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Sci. Rep. 2016, 6, 28101. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Chen, X.; Lombardi, R.; Willerson, J.T.; Marian, A.J. Knockdown of Plakophilin 2 Downregulates miR-184 Through CpG Hypermethylation and Suppression of the E2F1 Pathway and Leads to Enhanced Adipogenesis In Vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.B.; de Bakker, J.M.T.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Schlipp, A.; Schinner, C.; Spindler, V.; Vielmuth, F.; Gehmlich, K.; Syrris, P.; Mckenna, W.J.; Dendorfer, A.; Hartlieb, E.; Waschke, J. Desmoglein-2 interaction is crucial for cardiomyocyte cohesion and function. Cardiovasc. Res. 2014, 104, 245–257. [Google Scholar] [CrossRef]
- Valente, M.; Calabrese, F.; Thiene, G.; Angelini, A.; Basso, C.; Nava, A.; Rossi, L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am. J. Pathol. 1998, 152, 479–484. [Google Scholar]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 2006, 99, 646–655. [Google Scholar] [CrossRef]
- Zheng, G.; Jiang, C.; Li, Y.; Yang, D.; Ma, Y.; Zhang, B.; Li, X.; Zhang, P.; Hu, X.; Zhao, X.; et al. TMEM43-S358L mutation enhances NF-κB-TGFβ signal cascade in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Protein Cell 2019, 10, 104–119. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef]
- Campuzano, O.; Alcalde, M.; Iglesias, A.; Barahona-Dussault, C.; Sarquella-Brugada, G.; Benito, B.; Arzamendi, D.; Flores, J.; Leung, T.K.; Talajic, M.; et al. Arrhythmogenic right ventricular cardiomyopathy: Severe structural alterations are associated with inflammation. J. Clin. Pathol. 2012, 65, 1077–1083. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.-Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies with Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Bundgaard, H.; Edvardsen, T.; Eschen, O.; Gilljam, T.; Hansen, J.; Jensen, H.K.; Platonov, P.G.; Svensson, A.; Svendsen, J.H. Management of patients with Arrhythmogenic Right Ventricular Cardiomyopathy in the Nordic countries. Scand. Cardiovasc. J. 2015, 49, 299–307. [Google Scholar] [CrossRef]
- Gasperetti, A.; Cappelletto, C.; Carrick, R.; Targetti, M.; Tichnell, C.; Martino, A.; Murray, B.; Compagnucci, P.; Stolfo, D.; Bisson, J.; et al. Association of Premature Ventricular Contraction Burden on Serial Holter Monitoring with Arrhythmic Risk in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. JAMA Cardiol. 2022, 7, 378–385. [Google Scholar] [CrossRef]
- Prior, D.; La Gerche, A. Exercise and Arrhythmogenic Right Ventricular Cardiomyopathy. Heart Lung Circ. 2020, 29, 547–555. [Google Scholar] [CrossRef]
- Calkins, H.; Corrado, D.; Marcus, F. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2017, 136, 2068–2082. [Google Scholar] [CrossRef]
- McNally, E.; MacLeod, H.; Dellefave-Castillo, L. Arrhythmogenic Right Ventricular Cardiomyopathy Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1131/ (accessed on 21 November 2024).
- Gupta, R.; Tichnell, C.; Murray, B.; Rizzo, S.; Te Riele, A.; Tandri, H.; Judge, D.P.; Thiene, G.; Basso, C.; Calkins, H.; et al. Comparison of Features of Fatal Versus Nonfatal Cardiac Arrest in Patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Am. J. Cardiol. 2017, 120, 111–117. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, prognosis, and treatment. Heart 2000, 83, 588–595. [Google Scholar] [CrossRef]
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation 2005, 112, 3823–3832. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Frank, R.; Gallagher, J.J.; Reiter, M.J. Long-term follow-up in patients with arrhythmogenic right ventricular disease. Eur. Heart J. 1989, 10 (Suppl. D), 68–73. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Calkins, H.; Link, M.S.; Leoni, L.; Favale, S.; Bevilacqua, M.; Basso, C.; Ward, D.; Boriani, G.; Ricci, R.; et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 2010, 122, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Hulot, J.-S.; Jouven, X.; Empana, J.-P.; Frank, R.; Fontaine, G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004, 110, 1879–1884. [Google Scholar] [CrossRef]
- Baykan, A.; Olgar, Ş.; Argun, M.; Özyurt, A.; Pamukçu, Ö.; Üzüm, K.; Narin, N. Different clinical presentations of Naxos disease and Carvajal syndrome: Case series from a single tertiary center and review of the literature. Anatol. J. Cardiol. 2015, 15, 404–408. [Google Scholar] [CrossRef]
- Shah, S.N.; Umapathi, K.K.; Rout, P.; Horenstein, M.S.; Oliver, T.I. Arrhythmogenic Right Ventricular Cardiomyopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470378/ (accessed on 21 November 2024).
- Kottarthil, J.; Panuganti, P.; Jain, A.; Dalal, D.; Russell, S.D.; Judge, D.; Calkins, H.; Tandri, H. Abstract 12355: Role of Exercise Stress Testing in Arrhythmogenic Right Ventricular Dysplasia. Circulation 2010, 122 (Suppl. S21), A12355. [Google Scholar] [CrossRef]
- Lu, K.J.; Chen, J.X.C.; Profitis, K.; Kearney, L.G.; DeSilva, D.; Smith, G.; Ord, M.; Harberts, S.; Calafiore, P.; Jones, E.; et al. Right ventricular global longitudinal strain is an independent predictor of right ventricular function: A multimodality study of cardiac magnetic resonance imaging, real time three-dimensional echocardiography and speckle tracking echocardiography. Echocardiography 2015, 32, 966–974. [Google Scholar] [CrossRef]
- Wilkinson, J.C.; Colquitt, J.L.; Doan, T.T.; Liu, A.M.; Lilje, C.G.; Denfield, S.W.; Pignatelli, R.H.; Loar, R.W. Global Longitudinal Strain Analysis of the Single Right Ventricle: Leveling the Playing Field. J. Am. Soc. Echocardiogr. 2022, 35, 657–663. [Google Scholar] [CrossRef]
- Tolvaj, M.; Kovács, A.; Radu, N.; Cascella, A.; Muraru, D.; Lakatos, B.; Fábián, A.; Tokodi, M.; Tomaselli, M.; Gavazzoni, M.; et al. Significant Disagreement Between Conventional Parameters and 3D Echocardiography-Derived Ejection Fraction in the Detection of Right Ventricular Systolic Dysfunction and Its Association with Outcomes. J. Am. Soc. Echocardiogr. 2024, 37, 677–686. [Google Scholar] [CrossRef]
- Trimarchi, G.; Carerj, S.; Di Bella, G.; Manganaro, R.; Pizzino, F.; Restelli, D.; Pelaggi, G.; Lofrumento, F.; Licordari, R.; Taverna, G.; et al. Clinical Applications of Myocardial Work in Echocardiography: A Comprehensive Review. J. Cardiovasc. Echogr. 2024, 34, 99–113. [Google Scholar] [CrossRef]
- Krahn, A.D.; Wilde, A.A.M.; Calkins, H.; La Gerche, A.; Cadrin-Tourigny, J.; Roberts, J.D.; Han, H.-C. Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Clin. Electrophysiol. 2022, 8, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; James, C.A.; Tichnell, C.; Murray, B.; Gagarin, D.; Philips, B.; Dalal, D.; Tedford, R.; Russell, S.D.; Abraham, T.; et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J. Am. Coll. Cardiol. 2011, 58, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.M.; Srinivasan, N.T.; Rosmini, S.; Bulluck, H.; Orini, M.; Jenkins, S.; Pantazis, A.; McKenna, W.J.; Moon, J.C.; Lambiase, P.D.; et al. Electrical and Structural Substrate of Arrhythmogenic Right Ventricular Cardiomyopathy Determined Using Noninvasive Electrocardiographic Imaging and Late Gadolinium Magnetic Resonance Imaging. Circ. Arrhythm. Electrophysiol. 2017, 10, e005105. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Leoni, L.; Tokajuk, B.; Bauce, B.; Frigo, G.; Tarantini, G.; Napodano, M.; Turrini, P.; Ramondo, A.; et al. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2005, 111, 3042–3050. [Google Scholar] [CrossRef]
- Marra, M.P.; Leoni, L.; Bauce, B.; Corbetti, F.; Zorzi, A.; Migliore, F.; Silvano, M.; Rigato, I.; Tona, F.; Tarantini, G.; et al. Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: Comparison of 3D standard electroanatomical voltage mapping and contrast-enhanced cardiac magnetic resonance. Circ. Arrhythm. Electrophysiol. 2012, 5, 91–100. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Leoni, L.; Tokajuk, B.; Turrini, P.; Bauce, B.; Migliore, F.; Pavei, A.; Tarantini, G.; Napodano, M.; et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J. Am. Coll. Cardiol. 2008, 51, 731–739. [Google Scholar] [CrossRef]
- Corrado, D.; Anastasakis, A.; Basso, C.; Bauce, B.; Blomström-Lundqvist, C.; Bucciarelli-Ducci, C.; Cipriani, A.; De Asmundis, C.; Gandjbakhch, E.; Jiménez-Jáimez, J.; et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int. J. Cardiol. 2024, 395, 131447. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Basso, C.; Badano, L.P.; Bucciarelli-Ducci, C.; Cardim, N.; Gaemperli, O.; Galderisi, M.; Habib, G.; Knuuti, J.; Lancellotti, P.; et al. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyopathy-an expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 237–253. [Google Scholar] [CrossRef]
- Paul, M.; Wichter, T.; Kies, P.; Gerss, J.; Wollmann, C.; Rahbar, K.; Eckardt, L.; Breithardt, G.; Schober, O.; Schulze-Bahr, E.; et al. Cardiac sympathetic dysfunction in genotyped patients with arrhythmogenic right ventricular cardiomyopathy and risk of recurrent ventricular tachyarrhythmias. J. Nucl. Med. 2011, 52, 1559–1565. [Google Scholar] [CrossRef]
- Jacobson, A.F.; Senior, R.; Cerqueira, M.D.; Wong, N.D.; Thomas, G.S.; Lopez, V.A.; Agostini, D.; Weiland, F.; Chandna, H.; Narula, J.; et al. Myocardial iodine-123 meta-iodobenzylguanidine imaging and cardiac events in heart failure. Results of the prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study. J. Am. Coll. Cardiol. 2010, 55, 2212–2221. [Google Scholar] [CrossRef]
- te Riele, A.S.J.M.; James, C.A.; Groeneweg, J.A.; Sawant, A.C.; Kammers, K.; Murray, B.; Tichnell, C.; van der Heijden, J.F.; Judge, D.P.; Dooijes, D.; et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur. Heart J. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Sampognaro, J.R.; Gaine, S.P.; Sharma, A.; Tichnell, C.; Murray, B.; Shaik, Z.; Zimmerman, S.L.; James, C.A.; Gasperetti, A.; Calkins, H. Diagnostic pitfalls in patients referred for arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2023, 20, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Zorzi, A.; Cipriani, A.; Bauce, B.; Bariani, R.; Beffagna, G.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; et al. Evolving Diagnostic Criteria for Arrhythmogenic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e021987. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Zorzi, A.; Castelletti, S.; Pantasis, A.; Rigato, I.; Corrado, D.; Mckenna, W.; Lambiase, P.D. Electrocardiographic differentiation of idiopathic right ventricular outflow tract ectopy from early arrhythmogenic right ventricular cardiomyopathy. Europace 2017, 19, 622–628. [Google Scholar] [CrossRef]
- Brugada’, ’Josep; Diez’, ’Diego Perez How to Recognise and Manage Idiopathic Ventricular Tachycardia. Available online: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-8/How-to-recognise-and-manage-idiopathic-ventricular-tachycardia (accessed on 21 November 2024).
- Muser, D.; Santangeli, P.; Castro, S.A.; Casado Arroyo, R.; Maeda, S.; Benhayon, D.A.; Liuba, I.; Liang, J.J.; Sadek, M.M.; Chahal, A.; et al. Risk Stratification of Patients with Apparently Idiopathic Premature Ventricular Contractions: A Multicenter International CMR Registry. JACC Clin. Electrophysiol. 2020, 6, 722–735. [Google Scholar] [CrossRef]
- Wang, W.; Tichnell, C.; Murray, B.A.; Agafonova, J.; Cadrin-Tourigny, J.; Chelko, S.; Tandri, H.; Calkins, H.; James, C.A. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. Europace 2020, 22, 1270–1278. [Google Scholar] [CrossRef]
- Wichter, T.; Borggrefe, M.; Haverkamp, W.; Chen, X.; Breithardt, G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992, 86, 29–37. [Google Scholar] [CrossRef]
- Marcus, G.M.; Glidden, D.V.; Polonsky, B.; Zareba, W.; Smith, L.M.; Cannom, D.S.; Estes, N.A.M.; Marcus, F.; Scheinman, M.M. Efficacy of Antiarrhythmic Drugs in Arrhythmogenic Right Ventricular Cardiomyopathy: A Report from the North American ARVC Registry. J. Am. Coll. Cardiol. 2009, 54, 609–615. [Google Scholar] [CrossRef]
- Shen, L.-S.; Liu, L.-M.; Zheng, L.-H.; Hu, F.; Hu, Z.-C.; Liu, S.-Y.; Guo, J.-R.; Bhagat, K.K.; Yao, Y. Ablation strategies for arrhythmogenic right ventricular cardiomyopathy: A systematic review and meta-analysis. J. Geriatr. Cardiol. 2020, 17, 694–703. [Google Scholar] [CrossRef]
- Mathew, S.; Saguner, A.M.; Schenker, N.; Kaiser, L.; Zhang, P.; Yashuiro, Y.; Lemes, C.; Fink, T.; Maurer, T.; Santoro, F.; et al. Catheter Ablation of Ventricular Tachycardia in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Sequential Approach. J. Am. Heart Assoc. 2019, 8, e010365. [Google Scholar] [CrossRef] [PubMed]
- Gaine, S.P.; Calkins, H. Antiarrhythmic Drug Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy. Biomedicines 2023, 11, 1213. [Google Scholar] [CrossRef] [PubMed]
- Petruescu, L.; Lebreton, G.; Coutance, G.; Maupain, C.; Fressart, V.; Badenco, N.; Waintraub, X.; Duthoit, G.; Laredo, M.; Himbert, C.; et al. Clinical course of arrhythmogenic right ventricular cardiomyopathy with end-stage heart failure and outcome after heart transplantation. Arch. Cardiovasc. Dis. 2023, 116, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.; Scheel, P.; Etchill, E.; Fraser, C.D.; Suarez-Pierre, A.; Hsu, S.; Wittstein, I.S.; Kasper, E.K.; Florido, R.; Tandri, H.; et al. Heart transplantation outcomes in arrhythmogenic right ventricular cardiomyopathy: A contemporary national analysis. ESC Heart Fail. 2022, 9, 988–997. [Google Scholar] [CrossRef]
- Malik, N.; Mukherjee, M.; Wu, K.C.; Zimmerman, S.L.; Zhan, J.; Calkins, H.; James, C.A.; Gilotra, N.A.; Sheikh, F.H.; Tandri, H.; et al. Multimodality Imaging in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Imaging 2022, 15, e013725. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Location |
|---|---|---|
| JUP | Plakoglobin | Desmosome |
| DSP | Desmoplakin | Desmosome |
| PKP2 | Plakophilin 2 | Desmosome |
| DSC2 | Desmocollin 2 | Desmosome |
| DSG2 | Desmoglein 2 | Desmosome |
| SNC5A | NAv1.5 sodium channel | Cardiac cell membrane |
| TTN | Titin protein | Sarcomere |
| CTNNA3 | alpha-T catenin 3 | Intercalated disk |
| CDH2 | N-cadherin 2 | Intercalated disk |
| TJP1 | Tight junction protein 1 | Intercalated disk |
| LMNA | Lamin A/B | Nuclear envelope |
| TMEM43 | Luma | Nuclear envelope |
| PLN | Phospholamban | Sarcoplasmic reticulum |
| RYR2 | Ryanodine 2 receptors | Sarcoplasmic reticulum |
| DES | Desmin | Intermediate filaments |
| TGFB3 | Transforming growth factor beta 2 | Cytokine |
| Category | 2010 International Task Force ARVC Criteria | 2024 European Task Force ARVC Criteria |
|---|---|---|
| Morpho-Functional Ventricular Abnormalities | Major Echo
Echo
| By Echocardiography, Cardiac MRI, or Angiography Major
|
| Structural Alterations | Major
| Major
|
| Repolarization Abnormalities | Major
| Major
|
| Depolarization Abnormalities | Major
| Minor
|
| Ventricular Arrhythmias | Major
| Major
|
| Familial History | Major
| |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaikh, T.; Nguyen, D.; Dugal, J.K.; DiCaro, M.V.; Yee, B.; Houshmand, N.; Lei, K.; Namazi, A. Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2025, 12, 71. https://doi.org/10.3390/jcdd12020071
Shaikh T, Nguyen D, Dugal JK, DiCaro MV, Yee B, Houshmand N, Lei K, Namazi A. Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review. Journal of Cardiovascular Development and Disease. 2025; 12(2):71. https://doi.org/10.3390/jcdd12020071
Chicago/Turabian StyleShaikh, Taha, Darren Nguyen, Jasmine K. Dugal, Michael V. DiCaro, Brianna Yee, Nazanin Houshmand, KaChon Lei, and Ali Namazi. 2025. "Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review" Journal of Cardiovascular Development and Disease 12, no. 2: 71. https://doi.org/10.3390/jcdd12020071
APA StyleShaikh, T., Nguyen, D., Dugal, J. K., DiCaro, M. V., Yee, B., Houshmand, N., Lei, K., & Namazi, A. (2025). Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review. Journal of Cardiovascular Development and Disease, 12(2), 71. https://doi.org/10.3390/jcdd12020071