
Exploring the Current Status of Risk Stratification in Hypertrophic Cardiomyopathy: From Risk Models to Promising Techniques

 , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Current Risk Stratification Models
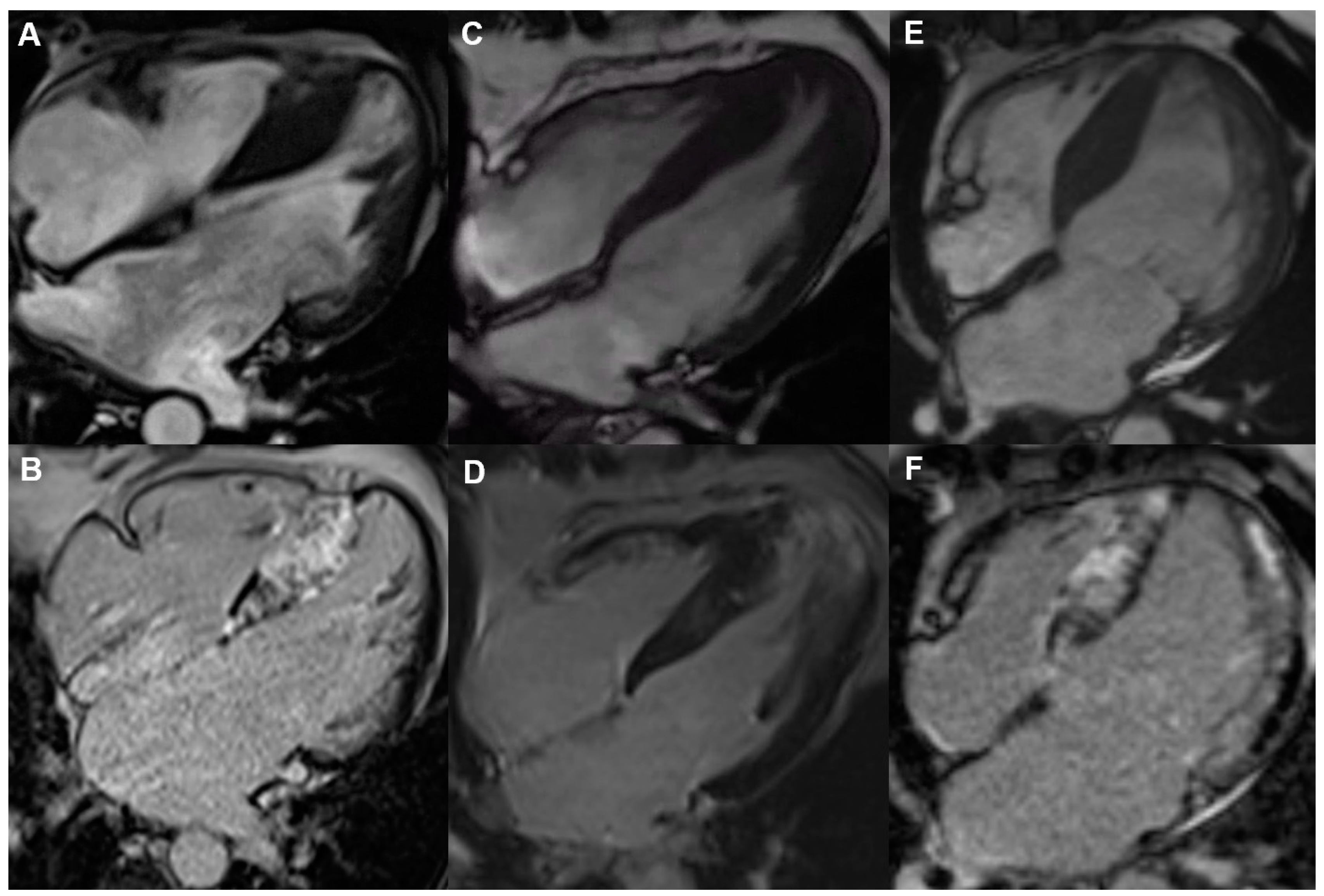
3. Late Gadolinium Enhancement in Cardiac Magnetic Resonance
4. Presence of a Left Ventricular Apical Aneurysm
5. Left Ventricle Systolic Dysfunction
6. Genetic Findings as Modifiers of Risk
7. Electrophysiological Testing
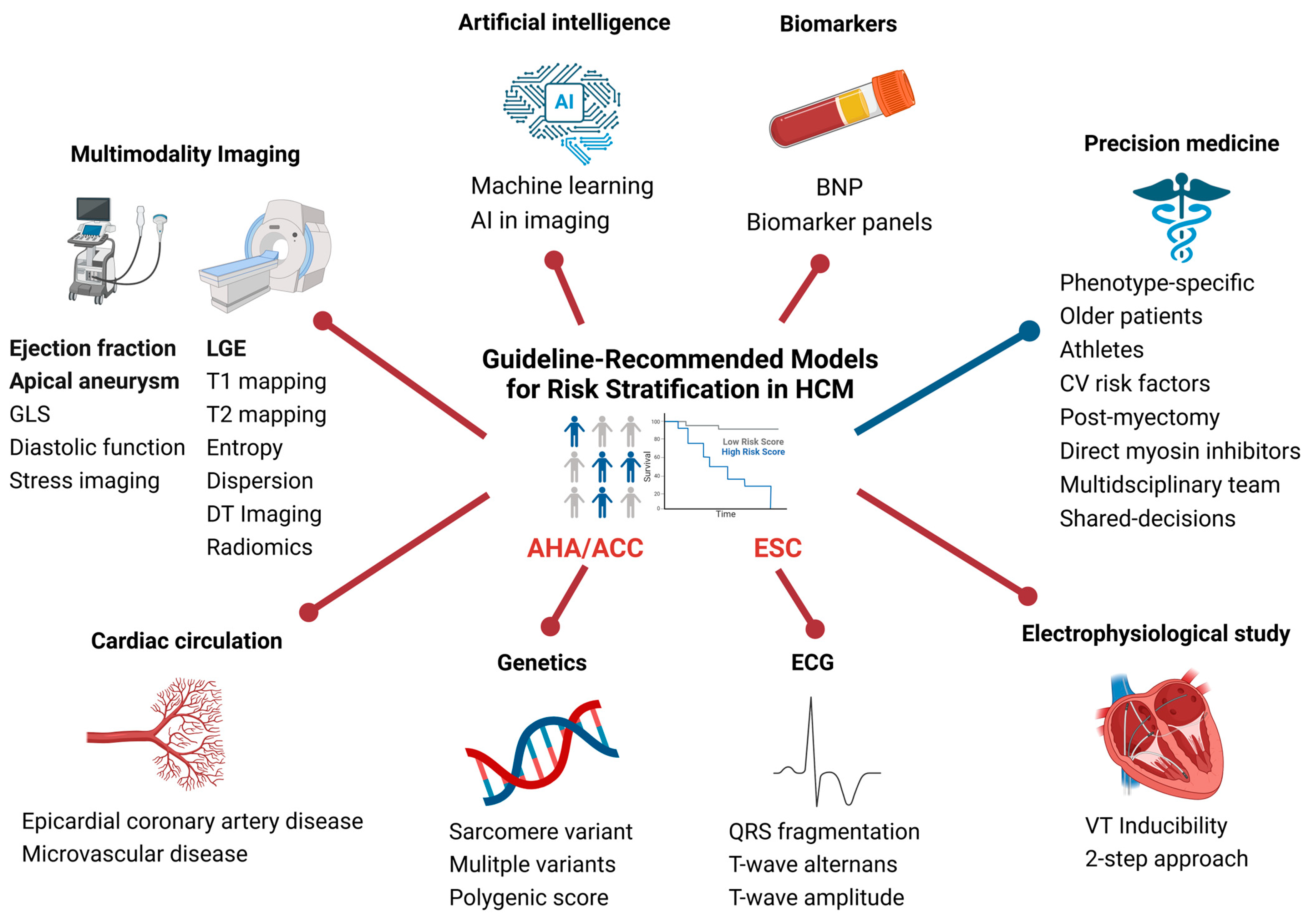
8. Novel Approaches to Risk Stratification in Hypertrophic Cardiomyopathy
8.1. Sub-Phenotype-Specific Approach to Risk Stratification
8.2. Multimodality Imaging
8.3. Artificial Intelligence
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | American College of Cardiology |
| AHA | American Heart Association |
| CMR | cardiac magnetic resonance |
| EP | electrophysiological |
| ESC | European Society of Cardiology |
| GWAS | genome-wide association studies |
| HCM | hypertrophic cardiomyopathy |
| ICD | implantable cardioverter defibrillator |
| LGE | late gadolinium enhancement |
| LVEF | left ventricular ejection fraction |
| LVH | left ventricular hypertrophy |
| LVMI | left ventricular mass index |
| LVOT | left ventricular outflow tract |
| LVSD | left ventricular systolic dysfunction |
| MWT | maximum wall thickness |
| NSVT | non-sustained ventricular tachycardia |
| PVS | programmed ventricular stimulation |
| SCD | sudden cardiac death |
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; De Boer, R.A.; et al. 2023 ESC Guidelines for the Management of Cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A Novel Clinical Risk Prediction Model for Sudden Cardiac Death in Hypertrophic Cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Bosman, L.P.; Nielsen Gerlach, C.L.; Cadrin-Tourigny, J.; Orgeron, G.; Tichnell, C.; Murray, B.; Bourfiss, M.; van der Heijden, J.F.; Yap, S.-C.; Zeppenfeld, K.; et al. Comparing Clinical Performance of Current Implantable Cardioverter-Defibrillator Implantation Recommendations in Arrhythmogenic Right Ventricular Cardiomyopathy. EP Eur. 2022, 24, 296–305. [Google Scholar] [CrossRef]
- Massera, D.; Sherrid, M.V.; Maron, M.S.; Rowin, E.J.; Maron, B.J. How Common Is Hypertrophic Cardiomyopathy… Really?: Disease Prevalence Revisited 27 Years after CARDIA. Int. J. Cardiol. 2023, 382, 64–67. [Google Scholar] [CrossRef]
- Abdelfattah, O.M.; Martinez, M.; Sayed, A.; ElRefaei, M.; Abushouk, A.I.; Hassan, A.; Masri, A.; Winters, S.L.; Kapadia, S.R.; Maron, B.J.; et al. Temporal and Global Trends of the Incidence of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. JACC Clin. Electrophysiol. 2022, 8, 1417–1427. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Papadakis, M.; Tanzarella, G.; Dhutia, H.; Miles, C.; Tome, M.; Behr, E.R.; Sharma, S.; Sheppard, M.N. Sudden Death Can Be the First Manifestation of Hypertrophic Cardiomyopathy. JACC Clin. Electrophysiol. 2019, 5, 252–254. [Google Scholar] [CrossRef]
- Maron, M.S.; Rowin, E.J.; Maron, B.J. The Paradigm of Sudden Death Prevention in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2024, 212, S64–S76. [Google Scholar] [CrossRef]
- Zörner, C.R.; Pallisgaard, J.; Schjerning, A.-M.; Jensen, M.K.; Tønnesen, J.; Da Riis-Vestergaard, L.; Middelfart, C.; Rasmussen, P.V.; Gislason, G.; Hansen, M.L. Temporal Trends of Hypertrophic Cardiomyopathy in Denmark: A Nationwide Retrospective Cohort Study. BMJ Open 2023, 13, e074010. [Google Scholar] [CrossRef]
- Maron, B.J.; McKenna, W.J.; Danielson, G.K.; Kappenberger, L.J.; Kuhn, H.J.; Seidman, C.E.; Shah, P.M.; Spencer, W.H., III; Spirito, P.; Ten Cate, F.J.; et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur. Heart J. 2003, 24, 1965–1991. [Google Scholar] [CrossRef]
- Writing Committee Members; Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef] [PubMed]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. Circulation 2011, 124, e783–e831. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Developed by the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- O’Mahony, C.; Jichi, F.; Ommen, S.R.; Christiaans, I.; Arbustini, E.; Garcia-Pavia, P.; Cecchi, F.; Olivotto, I.; Kitaoka, H.; Gotsman, I.; et al. International External Validation Study of the 2014 European Society of Cardiology Guidelines on Sudden Cardiac Death Prevention in Hypertrophic Cardiomyopathy (EVIDENCE-HCM). Circulation 2018, 137, 1015–1023. [Google Scholar] [CrossRef]
- Udelson, J.E. Evaluating and Reducing the Risk of Sudden Death in Hypertrophic Cardiomyopathy. Circulation 2019, 139, 727–729. [Google Scholar] [CrossRef]
- Vakrou, S.; Vlachopoulos, C.; Gatzoulis, K.A. Risk Stratification for Primary Prevention of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Arq. Bras. Cardiol. 2021, 117, 157–159. [Google Scholar] [CrossRef]
- Lazaros, G.; Antonopoulos, A.S.; Tousoulis, D. Risk Stratification in Hypertrophic Cardiomyopathy. J. Cardiovasc. Med. 2020, 21, 435. [Google Scholar] [CrossRef]
- Nauffal, V.; Marstrand, P.; Han, L.; Parikh, V.N.; Helms, A.S.; Ingles, J.; Jacoby, D.; Lakdawala, N.K.; Kapur, S.; Michels, M.; et al. Worldwide Differences in Primary Prevention Implantable Cardioverter Defibrillator Utilization and Outcomes in Hypertrophic Cardiomyopathy. Eur. Heart J. 2021, 42, 3932–3944. [Google Scholar] [CrossRef]
- Spirito, P.; Autore, C.; Formisano, F.; Assenza, G.E.; Biagini, E.; Haas, T.S.; Bongioanni, S.; Semsarian, C.; Devoto, E.; Musumeci, B.; et al. Risk of Sudden Death and Outcome in Patients with Hypertrophic Cardiomyopathy with Benign Presentation and Without Risk Factors. Am. J. Cardiol. 2014, 113, 1550–1555. [Google Scholar] [CrossRef]
- Rowin, E.J.; Burrows, A.; Madias, C.; Estes, N.A.M.; Link, M.S.; Maron, M.S.; Maron, B.J. Long-Term Outcome in High-Risk Patients with Hypertrophic Cardiomyopathy After Primary Prevention Defibrillator Implants. Circ. Arrhythm. Electrophysiol. 2020, 13, e008123. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Rowin, E.J.; Wessler, B.S.; Mooney, P.J.; Fatima, A.; Patel, P.; Koethe, B.C.; Romashko, M.; Link, M.S.; Maron, B.J. Enhanced American College of Cardiology/American Heart Association Strategy for Prevention of Sudden Cardiac Death in High-Risk Patients with Hypertrophic Cardiomyopathy. JAMA Cardiol. 2019, 4, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Zegkos, T.; Tziomalos, G.; Parcharidou, D.; Ntelios, D.; Papanastasiou, C.A.; Karagiannidis, E.; Gossios, T.; Rouskas, P.; Katranas, S.; Paraskevaidis, S.; et al. Validation of the New American College of Cardiology/American Heart Association Guidelines for the Risk Stratification of Sudden Cardiac Death in a Large Mediterranean Cohort with Hypertrophic Cardiomyopathy. Hellenic J. Cardiol. 2022, 63, 15–21. [Google Scholar] [CrossRef]
- Leong, K.M.W.; Chow, J.-J.; Ng, F.S.; Falaschetti, E.; Qureshi, N.; Koa-Wing, M.; Linton, N.W.F.; Whinnett, Z.I.; Lefroy, D.C.; Davies, D.W.; et al. Comparison of the Prognostic Usefulness of the European Society of Cardiology and American Heart Association/American College of Cardiology Foundation Risk Stratification Systems for Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 121, 349–355. [Google Scholar] [CrossRef]
- Ruivo, C.; Montenegro Sá, F.; Correia, J.; Belo, A.; Loureiro, M.F.; Morais, J. The SHIFT Model Combines Clinical, Electrocardiographic and Echocardiographic Parameters to Predict Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Rev. Port. Cardiol. 2019, 38, 847–853. [Google Scholar] [CrossRef]
- Kitaoka, H.; Tsutsui, H.; Kubo, T.; Ide, T.; Chikamori, T.; Fukuda, K.; Fujino, N.; Higo, T.; Isobe, M.; Kamiya, C.; et al. JCS/JHFS 2018 Guideline on the Diagnosis and Treatment of Cardiomyopathies. Circ. J. 2021, 85, 1590–1689. [Google Scholar] [CrossRef]
- Pelliccia, F.; Alfieri, O.; Calabrò, P.; Cecchi, F.; Ferrazzi, P.; Gragnano, F.; Kaski, J.P.; Limongelli, G.; Maron, M.; Rapezzi, C.; et al. Multidisciplinary Evaluation and Management of Obstructive Hypertrophic Cardiomyopathy in 2020: Towards the HCM Heart Team. Int. J. Cardiol. 2020, 304, 86–92. [Google Scholar] [CrossRef]
- Spirito, P.; Autore, C.; Rapezzi, C.; Bernabò, P.; Badagliacca, R.; Maron, M.S.; Bongioanni, S.; Coccolo, F.; Estes, N.A.M.; Barillà, C.S.; et al. Syncope and Risk of Sudden Death in Hypertrophic Cardiomyopathy. Circulation 2009, 119, 1703–1710. [Google Scholar] [CrossRef]
- Friedman, P.; Murgatroyd, F.; Boersma, L.V.A.; Manlucu, J.; O’Donnell, D.; Knight, B.P.; Clémenty, N.; Leclercq, C.; Amin, A.; Merkely, B.P.; et al. Efficacy and Safety of an Extravascular Implantable Cardioverter–Defibrillator. N. Engl. J. Med. 2022, 387, 1292–1302. [Google Scholar] [CrossRef]
- Francia, P.; Ziacchi, M.; Adduci, C.; Ammendola, E.; Pieragnoli, P.; De Filippo, P.; Rapacciuolo, A.; Rella, V.; Migliore, F.; Viani, S.; et al. Clinical Course of Hypertrophic Cardiomyopathy Patients Implanted with a Transvenous or Subcutaneous Defibrillator. EP Eur. 2023, 25, euad270. [Google Scholar] [CrossRef]
- Raiker, N.; Vullaganti, S.; Collins, J.D.; Allen, B.D.; Choudhury, L. Myocardial Tissue Characterization by Gadolinium-Enhanced Cardiac Magnetic Resonance Imaging for Risk Stratification of Adverse Events in Hypertrophic Cardiomyopathy. Int. J. Cardiovasc. Imaging 2020, 36, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Kariki, O.; Antoniou, C.-K.; Mavrogeni, S.; Gatzoulis, K.A. Updating the Risk Stratification for Sudden Cardiac Death in Cardiomyopathies: The Evolving Role of Cardiac Magnetic Resonance Imaging. An Approach for the Electrophysiologist. Diagnostics 2020, 10, 541. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S. Clinical Utility of Cardiovascular Magnetic Resonance in Hypertrophic Cardiomyopathy. J. Cardiovasc. Magn. Reson. 2012, 14, 7. [Google Scholar] [CrossRef]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402. [Google Scholar] [CrossRef]
- Adabag, A.S.; Maron, B.J.; Appelbaum, E.; Harrigan, C.J.; Buros, J.L.; Gibson, C.M.; Lesser, J.R.; Hanna, C.A.; Udelson, J.E.; Manning, W.J.; et al. Occurrence and Frequency of Arrhythmias in Hypertrophic Cardiomyopathy in Relation to Delayed Enhancement on Cardiovascular Magnetic Resonance. J. Am. Coll. Cardiol. 2008, 51, 1369–1374. [Google Scholar] [CrossRef]
- Choudhury, L.; Mahrholdt, H.; Wagner, A.; Choi, K.M.; Elliott, M.D.; Klocke, F.J.; Bonow, R.O.; Judd, R.M.; Kim, R.J. Myocardial Scarring in Asymptomatic or Mildly Symptomatic Patients with Hypertrophic Cardiomyopathy. JACC 2002, 40, 2156–2164. [Google Scholar] [CrossRef]
- Ismail, T.F.; Jabbour, A.; Gulati, A.; Mallorie, A.; Raza, S.; Cowling, T.E.; Das, B.; Khwaja, J.; Alpendurada, F.D.; Wage, R.; et al. Role of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in the Risk Stratification of Hypertrophic Cardiomyopathy. Heart Br. Card. Soc. 2014, 100, 1851–1858. [Google Scholar] [CrossRef]
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Pencina, M.J.; Assenza, G.E.; Haas, T.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; et al. Prognostic Value of Quantitative Contrast-Enhanced Cardiovascular Magnetic Resonance for the Evaluation of Sudden Death Risk in Patients with Hypertrophic Cardiomyopathy. Circulation 2014, 130, 484–495. [Google Scholar] [CrossRef]
- Mentias, A.; Raeisi-Giglou, P.; Smedira, N.G.; Feng, K.; Sato, K.; Wazni, O.; Kanj, M.; Flamm, S.D.; Thamilarasan, M.; Popovic, Z.B.; et al. Late Gadolinium Enhancement in Patients with Hypertrophic Cardiomyopathy and Preserved Systolic Function. J. Am. Coll. Cardiol. 2018, 72, 857–870. [Google Scholar] [CrossRef]
- Greulich, S.; Seitz, A.; Herter, D.; Günther, F.; Probst, S.; Bekeredjian, R.; Gawaz, M.; Sechtem, U.; Mahrholdt, H. Long-Term Risk of Sudden Cardiac Death in Hypertrophic Cardiomyopathy: A Cardiac Magnetic Resonance Outcome Study. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 732–741. [Google Scholar] [CrossRef]
- Arsenos, P.; Tsioufis, K.; Gatzoulis, K.A. Programmed Ventricular Stimulation for Risk Stratification in Hypertrophic Cardiomyopathy Patients. Eur. Heart J. 2024, 45, 2341–2342. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, S.; Ma, X.; Zhao, K.; Yang, K.; Yu, S.; Yin, G.; Dong, Z.; Song, Y.; Cui, C.; et al. Assessment of Late Gadolinium Enhancement in Hypertrophic Cardiomyopathy Improves Risk Stratification Based on Current Guidelines. Eur. Heart J. 2023, 44, 4781–4792. [Google Scholar] [CrossRef] [PubMed]
- Kiaos, A.; Daskalopoulos, G.N.; Kamperidis, V.; Ziakas, A.; Efthimiadis, G.; Karamitsos, T.D. Quantitative Late Gadolinium Enhancement Cardiac Magnetic Resonance and Sudden Death in Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2024, 17, 489–497. [Google Scholar] [CrossRef]
- Habib, M.; Adler, A.; Fardfini, K.; Hoss, S.; Hanneman, K.; Rowin, E.J.; Maron, M.S.; Maron, B.J.; Rakowski, H.; Chan, R.H. Progression of Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Cardiac Magnetic Resonance Study. JACC Cardiovasc. Imaging 2021, 14, 947–958. [Google Scholar] [CrossRef]
- Aquaro, G.D.; Todiere, G.; Barison, A.; Grigoratos, C.; Parisella, M.L.; Adami, M.; Grilli, G.; Pagura, L.; Faggioni, L.; Cioni, D.; et al. Prognostic Role of the Progression of Late Gadolinium Enhancement in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2024, 211, 199–208. [Google Scholar] [CrossRef]
- Klopotowski, M.; Kukula, K.; Malek, L.A.; Spiewak, M.; Polanska-Skrzypczyk, M.; Jamiolkowski, J.; Dabrowski, M.; Baranowski, R.; Klisiewicz, A.; Kusmierczyk, M.; et al. The Value of Cardiac Magnetic Resonance and Distribution of Late Gadolinium Enhancement for Risk Stratification of Sudden Cardiac Death in Patients with Hypertrophic Cardiomyopathy. J. Cardiol. 2016, 68, 49–56. [Google Scholar] [CrossRef]
- Bravo, P.E.; Luo, H.-C.; Pozios, I.; Zimmerman, S.L.; Corona-Villalobos, C.P.; Sorensen, L.; Kamel, I.R.; Bluemke, D.A.; Wahl, R.L.; Abraham, M.R.; et al. Late Gadolinium Enhancement Confined to the Right Ventricular Insertion Points in Hypertrophic Cardiomyopathy: An Intermediate Stage Phenotype? Eur. Heart J. Cardiovasc. Imaging 2016, 17, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, E.; Maron, B.J.; Adabag, S.; Hauser, T.H.; Lesser, J.R.; Haas, T.S.; Riley, A.B.; Harrigan, C.J.; Delling, F.N.; Udelson, J.E.; et al. Intermediate-Signal-Intensity Late Gadolinium Enhancement Predicts Ventricular Tachyarrhythmias in Patients with Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2012, 5, 78–85. [Google Scholar] [CrossRef]
- Aquaro, G.D.; Grigoratos, C.; Bracco, A.; Proclemer, A.; Todiere, G.; Martini, N.; Habtemicael, Y.G.; Carerj, S.; Sinagra, G.; Di Bella, G. Late Gadolinium Enhancement–Dispersion Mapping: A New Magnetic Resonance Imaging Technique to Assess Prognosis in Patients with Hypertrophic Cardiomyopathy and Low-Intermediate 5-Year Risk of Sudden Death. Circ. Cardiovasc. Imaging 2020, 13, e010489. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, B.J.; Haas, T.S.; Garberich, R.F.; Wang, W.; Link, M.S.; Maron, M.S. Hypertrophic Cardiomyopathy with Left Ventricular Apical Aneurysm. J. Am. Coll. Cardiol. 2017, 69, 761–773. [Google Scholar] [CrossRef]
- Lee, D.Z.J.; Montazeri, M.; Bataiosu, R.; Hoss, S.; Adler, A.; Nguyen, E.T.; Rakowski, H.; Chan, R.H. Clinical Characteristics and Prognostic Importance of Left Ventricular Apical Aneurysms in Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2022, 15, 1696–1711. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Finley, J.J.; Bos, J.M.; Hauser, T.H.; Manning, W.J.; Haas, T.S.; Lesser, J.R.; Udelson, J.E.; Ackerman, M.J.; Maron, B.J. Prevalence, Clinical Significance, and Natural History of Left Ventricular Apical Aneurysms in Hypertrophic Cardiomyopathy. Circulation 2008, 118, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Kolm, P.; Ho, C.Y.; Kwong, R.Y.; Desai, M.Y.; Dolman, S.F.; Appelbaum, E.; Desvigne-Nickens, P.; DiMarco, J.P.; Friedrich, M.G.; et al. Distinct Subgroups in Hypertrophic Cardiomyopathy in the NHLBI HCM Registry. J. Am. Coll. Cardiol. 2019, 74, 2333–2345. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Bernard, S.; Tripathi, N.; Patel, Y.; Modi, V.; Axel, L.; Talebi, S.; Ghoshhajra, B.B.; Sanborn, D.Y.; Saric, M.; et al. Apical Aneurysms and Mid–Left Ventricular Obstruction in Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2023, 16, 591–605. [Google Scholar] [CrossRef]
- Hughes, R.K.; Augusto, J.B.; Knott, K.; Davies, R.; Shiwani, H.; Seraphim, A.; Malcolmson, J.W.; Khoury, S.; Joy, G.; Mohiddin, S.; et al. Apical Ischemia Is a Universal Feature of Apical Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2023, 16, e014907. [Google Scholar] [CrossRef]
- Zenovich, A.G.; Lesser, J.R.; Hanna, C.A.; Maron, B.J. Identical Twins with Hypertrophic Cardiomyopathy and Apical Aneurysm. Am. J. Cardiol. 2006, 97, 1109. [Google Scholar] [CrossRef]
- Alfonso, F.; Frenneaux, M.P.; McKenna, W.J. Clinical Sustained Uniform Ventricular Tachycardia in Hypertrophic Cardiomyopathy: Association with Left Ventricular Apical Aneurysm. Heart 1989, 61, 178–181. [Google Scholar] [CrossRef]
- Papanastasiou, C.A.; Zegkos, T.; Karamitsos, T.D.; Rowin, E.J.; Maron, M.S.; Parcharidou, D.; Kokkinidis, D.G.; Karvounis, H.; Rimoldi, O.; Maron, B.J.; et al. Prognostic Role of Left Ventricular Apical Aneurysm in Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2021, 332, 127–132. [Google Scholar] [CrossRef]
- Rouskas, P.; Zegkos, T.; Ntelios, D.; Gossios, T.; Parcharidou, D.; Papanastasiou, C.A.; Karamitsos, T.; Vassilikos, V.; Kouskouras, K.; Efthimiadis, G.K. Prevalence, Characteristics, and Natural History of Apical Phenotype in a Large Cohort of Patients with Hypertrophic Cardiomyopathy. Hell. J. Cardiol. 2023, 73, 8–15. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.-Y.; Chen, X.-Y.; Wang, J.-X.; Li, L.; Yin, G.; Zheng, Y.-C.; Wei, M.-D.; Lu, M.-J.; Zhao, S.-H. Apical Hypertrophic Cardiomyopathy with Left Ventricular Apical Aneurysm: Prevalence, Cardiac Magnetic Resonance Characteristics, and Prognosis. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 1341–1350. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Massera, D.; Bernard, S.; Tripathi, N.; Patel, Y.; Modi, V.; Axel, L.; Talebi, S.; Saric, M.; Adlestein, E.; et al. Clinical Course and Treatment of Patients with Apical Aneurysms Due to Hypertrophic Cardiomyopathy. JACC Adv. 2024, 3, 101195. [Google Scholar] [CrossRef] [PubMed]
- Dilaveris, P.; Aggeli, C.; Synetos, A.; Skiadas, I.; Antoniou, C.; Tsiamis, E.; Gatzoulis, K.; Kallikazaros, I.; Tousoulis, D. Sustained Ventricular Tachycardia as a First Manifestation of Hypertrophic Cardiomyopathy with Mid-ventricular Obstruction and Apical Aneurysm in an Elderly Female Patient. Ann. Noninvasive Electrocardiol. 2016, 22, e12422. [Google Scholar] [CrossRef]
- Igarashi, M.; Nogami, A.; Kurosaki, K.; Hanaki, Y.; Komatsu, Y.; Fukamizu, S.; Morishima, I.; Kaitani, K.; Nishiuchi, S.; Talib, A.K.; et al. Radiofrequency Catheter Ablation of Ventricular Tachycardia in Patients with Hypertrophic Cardiomyopathy and Apical Aneurysm. JACC Clin. Electrophysiol. 2018, 4, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Maron, B.J.; Appelbaum, E.; Harrigan, C.J.; Salton, C.; Gibson, C.M.; Udelson, J.E.; O’Donnell, C.; Lesser, J.R.; Manning, W.J.; et al. Spectrum and Clinical Significance of Systolic Function and Myocardial Fibrosis Assessed by Cardiovascular Magnetic Resonance in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2010, 106, 261–267. [Google Scholar] [CrossRef]
- Wasserstrum, Y.; Larrañaga-Moreira, J.M.; Martinez-Veira, C.; Itelman, E.; Lotan, D.; Sabbag, A.; Kuperstein, R.; Peled, Y.; Freimark, D.; Barriales-Villa, R.; et al. Hypokinetic Hypertrophic Cardiomyopathy: Clinical Phenotype, Genetics, and Prognosis. ESC Heart Fail. 2022, 9, 2301–2312. [Google Scholar] [CrossRef]
- Marstrand, P.; Han, L.; Day, S.M.; Olivotto, I.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Wittekind, S.G.; Helms, A.; Saberi, S.; et al. Hypertrophic Cardiomyopathy with Left Ventricular Systolic Dysfunction. Circulation 2020, 141, 1371–1383. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, B.J.; Carrick, R.T.; Patel, P.P.; Koethe, B.; Wells, S.; Maron, M.S. Outcomes in Patients with Hypertrophic Cardiomyopathy and Left Ventricular Systolic Dysfunction. J. Am. Coll. Cardiol. 2020, 75, 3033–3043. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Kim, H.-K.; Hwang, I.-C.; Park, C.S.; Rhee, T.-M.; Lee, H.-J.; Park, J.-B.; Yoon, Y.E.; Lee, S.-P.; Cho, G.-Y.; et al. Prognosis of Patients with Hypertrophic Cardiomyopathy and Low-Normal Left Ventricular Ejection Fraction. Heart 2023, 109, 771–778. [Google Scholar] [CrossRef]
- Solomou, E.; Gatzoulis, K.A.; Skiadas, I.; Doundoulakis, I.; Arsenos, P.; Dilaveris, P.; Sideris, S.; Tousoulis, D. Upgrade to Cardiac Resynchronization Therapy Difibrillator Device of a Pacemaker-Dependent Patient with End-Stage Hypertrophic Cardiomyopathy. Hellenic J. Cardiol. 2020, 61, 65–67. [Google Scholar] [CrossRef]
- Lorenzini, M.; Norrish, G.; Field, E.; Ochoa, J.P.; Cicerchia, M.; Akhtar, M.M.; Syrris, P.; Lopes, L.R.; Kaski, J.P.; Elliott, P.M. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. J. Am. Coll. Cardiol. 2020, 76, 550–559. [Google Scholar] [CrossRef]
- Lopes, L.R.; Barbosa, P.; Torrado, M.; Quinn, E.; Merino, A.; Ochoa, J.P.; Jager, J.; Futema, M.; Carmo-Fonseca, M.; Monserrat, L.; et al. Cryptic Splice-Altering Variants in MYBPC3 Are a Prevalent Cause of Hypertrophic Cardiomyopathy. Circ. Genomic Precis. Med. 2020, 13, e002905. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.; Bagnall, R.D.; Lam, L.; Semsarian, C.; Ingles, J. Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet. 2017, 10, e001666. [Google Scholar] [CrossRef]
- Watkins, H.; Rosenzweig, A.; Hwang, D.-S.; Levi, T.; McKenna, W.; Seidman, C.E.; Seidman, J.G. Characteristics and Prognostic Implications of Myosin Missense Mutations in Familial Hypertrophic Cardiomyopathy. N. Engl. J. Med. 1992, 326, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; McKenna, W.J.; Thierfelder, L.; Suk, H.J.; Anan, R.; O’Donoghue, A.; Spirito, P.; Matsumori, A.; Moravec, C.S.; Seidman, J.G.; et al. Mutations in the Genes for Cardiac Troponin T and α-Tropomyosin in Hypertrophic Cardiomyopathy. N. Engl. J. Med. 1995, 332, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Dubourg, O.; Desnos, M.; Bennaceur, M.; Carrier, L.; Camproux, A.-C.; Isnard, R.; Hagege, A.; Langlard, J.M.; Bonne, G.; et al. Clinical Features and Prognostic Implications of Familial Hypertrophic Cardiomyopathy Related to the Cardiac Myosin-Binding Protein C Gene. Circulation 1998, 97, 2230–2236. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Jaeger, M.A.; Ommen, S.R.; Will, M.L.; Gersh, B.J.; Tajik, A.J.; Ackerman, M.J. Comprehensive Analysis of the Beta-Myosin Heavy Chain Gene in 389 Unrelated Patients with Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 602–610. [Google Scholar] [CrossRef]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament Protein Gene Mutation Screening and Outcome of Patients with Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef]
- Pasquale, F.; Syrris, P.; Kaski, J.P.; Mogensen, J.; McKenna, W.J.; Elliott, P. Long-Term Outcomes in Hypertrophic Cardiomyopathy Caused by Mutations in the Cardiac Troponin T Gene. Circ. Cardiovasc. Genet. 2012, 5, 10–17. [Google Scholar] [CrossRef]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical Phenotype and Outcome of Hypertrophic Cardiomyopathy Associated with Thin-Filament Gene Mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel Genotype–Phenotype Associations Demonstrated by High-Throughput Sequencing in Patients with Hypertrophic Cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef]
- Lee, S.-P.; Ashley, E.A.; Homburger, J.; Caleshu, C.; Green, E.M.; Jacoby, D.; Colan, S.D.; Arteaga-Fernández, E.; Day, S.M.; Girolami, F.; et al. Incident Atrial Fibrillation Is Associated with MYH7 Sarcomeric Gene Variation in Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2018, 11, e005191. [Google Scholar] [CrossRef] [PubMed]
- Helms, A.S.; Thompson, A.D.; Glazier, A.A.; Hafeez, N.; Kabani, S.; Rodriguez, J.; Yob, J.M.; Woolcock, H.; Mazzarotto, F.; Lakdawala, N.K.; et al. Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients with Hypertrophic Cardiomyopathy. Circ. Genomic Precis. Med. 2020, 13, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Melendo-Viu, M.; Salguero-Bodes, R.; Valverde-Gómez, M.; Larrañaga-Moreira, J.M.; Barriales, R.; Díez-Lopez, C.; Freire, J.L.; Peña-Peña, M.L.; Pavia, P.G.; Ripoll, T.; et al. Hypertrophic Cardiomyopathy Due to Truncating Variants in Myosin Binding Protein C: A Spanish Cohort. Open Heart 2024, 11, e002891. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Ackerman, M.J. Mutation Type Is Not Clinically Useful in Predicting Prognosis in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2441–2450. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Bonaventura, J.; Rowin, E.J.; Chan, R.H.; Chin, M.T.; Puchnerova, V.; Polakova, E.; Macek, M.; Votypka, P.; Batorsky, R.; Perera, G.; et al. Relationship Between Genotype Status and Clinical Outcome in Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e033565. [Google Scholar] [CrossRef]
- Biddinger, K.J.; Jurgens, S.J.; Maamari, D.; Gaziano, L.; Choi, S.H.; Morrill, V.N.; Halford, J.L.; Khera, A.V.; Lubitz, S.A.; Ellinor, P.T.; et al. Rare and Common Genetic Variation Underlying the Risk of Hypertrophic Cardiomyopathy in a National Biobank. JAMA Cardiol. 2022, 7, 715–722. [Google Scholar] [CrossRef]
- Ning, C.; Fan, L.; Jin, M.; Wang, W.; Hu, Z.; Cai, Y.; Chen, L.; Lu, Z.; Zhang, M.; Chen, C.; et al. Genome-Wide Association Analysis of Left Ventricular Imaging-Derived Phenotypes Identifies 72 Risk Loci and Yields Genetic Insights into Hypertrophic Cardiomyopathy. Nat. Commun. 2023, 14, 7900. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Zheng, S.L.; Jurgens, S.J.; McGurk, K.A.; Xu, X.; Grace, C.; Theotokis, P.I.; Buchan, R.J.; Francis, C.; de Marvao, A.; Curran, L.; et al. Evaluation of Polygenic Scores for Hypertrophic Cardiomyopathy in the General Population and across Clinical Settings. Nat. Genet. 2025, 57, 563–571. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Losi, M.-A.; Sheikh, N.; Laroche, C.; Charron, P.; Gimeno, J.; Kaski, J.P.; Maggioni, A.P.; Tavazzi, L.; Arbustini, E.; et al. Association between Common Cardiovascular Risk Factors and Clinical Phenotype in Patients with Hypertrophic Cardiomyopathy from the European Society of Cardiology (ESC) EurObservational Research Programme (EORP) Cardiomyopathy/Myocarditis Registry. Eur. Heart J.-Qual. Care Clin. Outcomes 2023, 9, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.M.; Schwartz, J.L.; Maron, B.J.; Tucker, E.; Rosing, D.R.; Josephson, M.E. Inducible Polymorphic Ventricular Tachycardia and Ventricular Fibrillation in a Subgroup of Patients with Hypertrophic Cardiomyopathy at High Risk for Sudden Death. J. Am. Coll. Cardiol. 1987, 10, 761–774. [Google Scholar] [CrossRef]
- Fananapazir, L.; Chang, A.C.; Epstein, S.E.; McAreavey, D. Prognostic Determinants in Hypertrophic Cardiomyopathy. Prospective Evaluation of a Therapeutic Strategy Based on Clinical, Holter, Hemodynamic, and Electrophysiological Findings. Circulation 1992, 86, 730–740. [Google Scholar] [CrossRef]
- Kuck, K.-H.; Kunze, K.-P.; Schluter, M.; Nienaber, C.A.; Costard, A. Programmed electrical stimulation in hypertrophic cardiomyopathy. Results in patients with and without cardiac arrest or syncope. Eur. Heart J. 1988, 9, 177–185. [Google Scholar] [CrossRef]
- Sanghvi, M.M.; Dhall, E.; Anwar, A.; Chahal, C.; O’Mahony, C.; Mohiddin, S.A.; Savvatis, K.; Ricci, F.; Munroe, P.B.; Petersen, S.E.; et al. Hypertrophic Cardiomyopathy Management: A Systematic Review of the Clinical Practice Guidelines and Recommendations. Eur. Heart J.-Qual. Care Clin. Outcomes 2025, qcae117. [Google Scholar] [CrossRef]
- Gatzoulis, K.A.; Georgopoulos, S.; Anastasakis, A.; Antoniou, C.-K.; Arsenos, P.; Tsiachris, D.; Dilaveris, P.; Sideris, S.; Tousoulis, D. Arrhythmic Risk Stratification in Hypertrophic Cardiomyopathy: Are We Missing Something? EP Eur. 2021, 23, 648–649. [Google Scholar] [CrossRef]
- Gatzoulis, K.A.; Georgopoulos, S.; Antoniou, C.-K.; Anastasakis, A.; Dilaveris, P.; Arsenos, P.; Sideris, S.; Tsiachris, D.; Archontakis, S.; Sotiropoulos, E.; et al. Programmed Ventricular Stimulation Predicts Arrhythmic Events and Survival in Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2018, 254, 175–181. [Google Scholar] [CrossRef]
- Jang, S.Y.; Kim, W.S.; Lee, S.-C. Natural History of Hypertrophic Cardiomyopathy in Korea: A Nationwide Population-Based Retrospective Cohort Study. J. Korean Med. Sci. 2024, 40, e61. [Google Scholar] [CrossRef]
- Wu, S.; Yang, L.; Sun, N.; Luo, X.; Li, P.; Wang, K.; Li, P.; Zhao, J.; Wang, Z.; Zhang, Q.; et al. Impact of Coronary Artery Disease in Patients with Hypertrophic Cardiomyopathy. Hellenic J. Cardiol. 2024, 77, 27–35. [Google Scholar] [CrossRef]
- Doundoulakis, I.; Tsiachris, D.; Kordalis, A.; Soulaidopoulos, S.; Arsenos, P.; Xintarakou, A.; Koliastasis, L.; Vlachakis, P.K.; Tsioufis, K.; Gatzoulis, K.A. Management of Patients with Unexplained Syncope and Bundle Branch Block: Predictive Factors of Recurrent Syncope. Cureus 2023, 15, e35827. [Google Scholar] [CrossRef] [PubMed]
- Arkolaki, E.G.; Simantirakis, E.N.; Chrysostomakis, S.I.; Vardas, P.E. Appropriate Management of Syncope in a Patient with Hypertrophic Cardiomyopathy: Rationale behind Long-Term Cardiac Rhythm Monitoring. Hellenic J. Cardiol. 2009, 50, 144–146. [Google Scholar] [PubMed]
- Tang, W.; Liu, M.; Li, J.; Chang, R.; Su, C.; Zhang, X.; Wang, L. The Risk of Ventricular Arrhythmias between Alcohol Septal Ablation and Septal Myectomy in Hypertrophic Cardiomyopathy: A Meta-Analysis on Septal Reduction Therapy. Rev. Cardiovasc. Med. 2022, 23, 391. [Google Scholar] [CrossRef] [PubMed]
- Saumarez, R.; Silberbauer, J.; Scannell, J.; Pytkowski, M.; Behr, E.R.; Betts, T.; Della Bella, P.; Peters, N.S. Should Lethal Arrhythmias in Hypertrophic Cardiomyopathy Be Predicted Using Non-Electrophysiological Methods? EP Eur. 2023, 25, euad045. [Google Scholar] [CrossRef]
- Elliott, P.M.; Poloniecki, J.; Dickie, S.; Sharma, S.; Monserrat, L.; Varnava, A.; Mahon, N.G.; McKenna, W.J. Sudden Death in Hypertrophic Cardiomyopathy: Identification of High Risk Patients. J. Am. Coll. Cardiol. 2000, 36, 2212–2218. [Google Scholar] [CrossRef]
- Saumarez, R.C.; Pytkowski, M.; Sterlinski, M.; Bourke, J.P.; Clague, J.R.; Cobbe, S.M.; Connelly, D.T.; Griffith, M.J.; McKeown, P.P.; McLeod, K.; et al. Paced Ventricular Electrogram Fractionation Predicts Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Eur. Heart J. 2008, 29, 1653–1661. [Google Scholar] [CrossRef]
- Fluechter, S.; Kuschyk, J.; Wolpert, C.; Doesch, C.; Veltmann, C.; Haghi, D.; Schoenberg, S.O.; Sueselbeck, T.; Germans, T.; Streitner, F.; et al. Extent of Late Gadolinium Enhancement Detected by Cardiovascular Magnetic Resonance Correlates with the Inducibility of Ventricular Tachyarrhythmia in Hypertrophic Cardiomyopathy. J. Cardiovasc. Magn. Reson. 2010, 12, 30. [Google Scholar] [CrossRef]
- Francia, P.; Falasconi, G.; Penela, D.; Viveros, D.; Alderete, J.; Saglietto, A.; Bellido, A.F.; Martí-Almor, J.; Franco-Ocaña, P.; Soto-Iglesias, D.; et al. Scar Architecture Affects the Electrophysiological Characteristics of Induced Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. EP Eur. 2024, 26, euae050. [Google Scholar] [CrossRef]
- Hajj-Ali, A.; Gaballa, A.; Akintoye, E.; Jadam, S.; Ramchand, J.; Xu, B.; Ospina, S.; Thamilarasan, M.; Smedira, N.G.; Popovic, Z.B.; et al. Long-Term Outcomes of Patients with Apical Hypertrophic Cardiomyopathy Utilizing a New Risk Score. JACC Adv. 2024, 3, 101235. [Google Scholar] [CrossRef]
- Tower-Rader, A.; Kramer, C.M.; Neubauer, S.; Nagueh, S.F.; Desai, M.Y. Multimodality Imaging in Hypertrophic Cardiomyopathy for Risk Stratification. Circ. Cardiovasc. Imaging 2020, 13, e009026. [Google Scholar] [CrossRef]
- Tower-Rader, A.; Mohananey, D.; To, A.; Lever, H.M.; Popovic, Z.B.; Desai, M.Y. Prognostic Value of Global Longitudinal Strain in Hypertrophic Cardiomyopathy: A Systematic Review of Existing Literature. JACC Cardiovasc. Imaging 2019, 12, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, H.K.; Lee, S.C.; Kim, J.; Park, J.B.; Hwang, I.C.; Choi, Y.J.; Lee, S.P.; Chang, S.A.; Lee, W.; et al. Supplementary Role of Left Ventricular Global Longitudinal Strain for Predicting Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Eur. Heart J.-Cardiovasc. Imaging 2022, 23, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Negri, F.; Muser, D.; Driussi, M.; Sanna, G.D.; Masè, M.; Cittar, M.; Poli, S.; De Bellis, A.; Fabris, E.; Puppato, M.; et al. Prognostic Role of Global Longitudinal Strain by Feature Tracking in Patients with Hypertrophic Cardiomyopathy: The STRAIN-HCM Study. Int. J. Cardiol. 2021, 345, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Alajmi, F.; Kang, M.; Dundas, J.; Haenel, A.; Parker, J.; Blanke, P.; Coghlan, F.; Khoo, J.K.; Bin Zaid, A.A.; Singh, A.; et al. Novel Magnetic Resonance Imaging Tools for Hypertrophic Cardiomyopathy Risk Stratification. Life 2024, 14, 200. [Google Scholar] [CrossRef]
- Zhao, X.; Jin, F.; Wang, J.; Zhao, X.; Wang, L.; Wei, H. Entropy of Left Ventricular Late Gadolinium Enhancement and Its Prognostic Value in Hypertrophic Cardiomyopathy a New CMR Assessment Method. Int. J. Cardiol. 2023, 373, 134–141. [Google Scholar] [CrossRef]
- Gu, Z.-Y.; Qian, Y.-F.; Chen, B.-H.; Wu, C.-W.; Zhao, L.; Xue, S.; Zhao, L.; Wu, L.-M.; Wang, Y.-Y. Late Gadolinium Enhancement Entropy as a New Measure of Myocardial Tissue Heterogeneity for Prediction of Adverse Cardiac Events in Patients with Hypertrophic Cardiomyopathy. Insights Imaging 2023, 14, 138. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Pu, L.; Qi, W.; Xu, Y.; Wan, K.; Zhu, Y.; Gkoutos, G.V.; Han, Y.; Chen, Y. The Prognostic Value of Left Ventricular Entropy From T1 Mapping in Patients with Hypertrophic Cardiomyopathy. JACC Asia 2024, 4, 389–399. [Google Scholar] [CrossRef]
- Dall’Armellina, E.; Ennis, D.B.; Axel, L.; Croisille, P.; Ferreira, P.F.; Gotschy, A.; Lohr, D.; Moulin, K.; Nguyen, C.T.; Nielles-Vallespin, S.; et al. Cardiac Diffusion-Weighted and Tensor Imaging: A Consensus Statement from the Special Interest Group of the Society for Cardiovascular Magnetic Resonance. J. Cardiovasc. Magn. Reson. 2025, 27, 101109. [Google Scholar] [CrossRef]
- Khalique, Z.; Ferreira, P.F.; Scott, A.D.; Nielles-Vallespin, S.; Firmin, D.N.; Pennell, D.J. Diffusion Tensor Cardiovascular Magnetic Resonance Imaging: A Clinical Perspective. JACC Cardiovasc. Imaging 2020, 13, 1235–1255. [Google Scholar] [CrossRef]
- Joy, G.; Kelly, C.I.; Webber, M.; Pierce, I.; Teh, I.; McGrath, L.; Velazquez, P.; Hughes, R.K.; Kotwal, H.; Das, A.; et al. Microstructural and Microvascular Phenotype of Sarcomere Mutation Carriers and Overt Hypertrophic Cardiomyopathy. Circulation 2023, 148, 808–818. [Google Scholar] [CrossRef]
- Das, A.; Kelly, C.; Teh, I.; Nguyen, C.; Brown, L.A.E.; Chowdhary, A.; Jex, N.; Thirunavukarasu, S.; Sharrack, N.; Gorecka, M.; et al. Phenotyping Hypertrophic Cardiomyopathy Using Cardiac Diffusion Magnetic Resonance Imaging: The Relationship between Microvascular Dysfunction and Microstructural Changes. Eur. Heart J.-Cardiovasc. Imaging 2022, 23, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Smole, T.; Žunkovič, B.; Pičulin, M.; Kokalj, E.; Robnik-Šikonja, M.; Kukar, M.; Fotiadis, D.I.; Pezoulas, V.C.; Tachos, N.S.; Barlocco, F.; et al. A Machine Learning-Based Risk Stratification Model for Ventricular Tachycardia and Heart Failure in Hypertrophic Cardiomyopathy. Comput. Biol. Med. 2021, 135, 104648. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.; Kpodonu, J.; Agu, E. Risk Stratification in Hypertrophic Cardiomyopathy. JACC Adv. 2023, 2, 100562. [Google Scholar] [CrossRef] [PubMed]
- Tsampras, T.; Karamanidou, T.; Papanastasiou, G.; Stavropoulos, T.G. Deep Learning for Cardiac Imaging: Focus on Myocardial Diseases, a Narrative Review. Hell. J. Cardiol. 2025, 81, 18–24. [Google Scholar] [CrossRef]
- Augusto, J.B.; Davies, R.H.; Bhuva, A.N.; Knott, K.D.; Seraphim, A.; Alfarih, M.; Lau, C.; Hughes, R.K.; Lopes, L.R.; Shiwani, H.; et al. Diagnosis and Risk Stratification in Hypertrophic Cardiomyopathy Using Machine Learning Wall Thickness Measurement: A Comparison with Human Test-Retest Performance. Lancet Digit. Health 2021, 3, e20–e28. [Google Scholar] [CrossRef]
- Xu, H.; Williams, S.E.; Williams, M.C.; Newby, D.E.; Taylor, J.; Neji, R.; Kunze, K.P.; Niederer, S.A.; Young, A.A. Deep Learning Estimation of Three-Dimensional Left Atrial Shape from Two-Chamber and Four-Chamber Cardiac Long Axis Views. Eur. Heart J.-Cardiovasc. Imaging 2023, 24, 607–615. [Google Scholar] [CrossRef]
- Sahota, M.; Saraskani, S.R.; Xu, H.; Li, L.; Majeed, A.W.; Hermida, U.; Neubauer, S.; Desai, M.; Weintraub, W.; Desvigne-Nickens, P.; et al. Machine Learning Evaluation of LV Outflow Obstruction in Hypertrophic Cardiomyopathy Using Three-Chamber Cardiovascular Magnetic Resonance. Int. J. Cardiovasc. Imaging 2022, 38, 2695–2705. [Google Scholar] [CrossRef]
- Carrick, R.T.; Ahamed, H.; Sung, E.; Maron, M.S.; Madias, C.; Avula, V.; Studley, R.; Bao, C.; Bokhari, N.; Quintana, E.; et al. Identification of High-Risk Imaging Features in Hypertrophic Cardiomyopathy Using Electrocardiography: A Deep-Learning Approach. Heart Rhythm 2024, 21, 1390–1397. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| ACC/ESC 2003 | ACC/AHA 2011 | ESC 2014 | AHA/ACC 2020 | ESC 2023 | AHA/ACC 2024 | |
|---|---|---|---|---|---|---|
| Risk assessment model | Major risk factors
| Major risk factors (≥1)
| HCM Risk-SCD prediction score of SCD at 5 years * ≥6% (Class IIa) ≥4–6% (Class IIb) <4% (Class IIb) | Major risk factors (≥1)
| HCM Risk-SCD prediction score of SCD at 5 years * ≥6% (Class IIa) ≥4–6% (Class IIb) <4% (Class IIb) & Risk modifiers | Major risk factors (≥1)
|
| NSVT | Considered major risk factor | In the presence of other SCD risk modifiers (Class IIa) | Included in risk score | May be considered if no major risk factors or decision uncertain particularly if frequent, long and fast (Class IIb) | Included in risk score. There is no evidence that the frequency, duration, or rate of NSVT influences risk | May be considered if no major risk factors or decision uncertain particularly if frequent, long and fast (Class IIb) |
| Abnormal BP response | Considered major risk factor | In the presence of other SCD risk modifiers (Class IIa) | Not recommended | Not recommended | Not recommended | Not recommended |
| LVOT obstruction gradient | <30 mmHg compatible with lower risk | Usefulness is unclear but might be considered in selected patients (Class IIb) | Included in risk score | No mention | Included in risk score | No mention |
| LV systolic function | No mention | No mention | No mention | Major risk factor (LVEF < 50%) (Class IIa) | An LVEF <50% is a clinical risk factor to be considered in patients in the low (to intermediate) risk category (Class IIb) | Major risk factor (LVEF < 50%) (Class IIa) |
| LV apical aneurysm | No mention | May warrant consideration though evidence is limited | No recommendation due to few data | Major risk factor (Class IIa) | Decisions should not be solely based on presence of apical aneurysm | Major risk factor (Class IIa) |
| Late gadolinium enhancement | No mention | Usefulness is unclear but might be considered in selected patients (Class IIb) | No recommendation due to few data | If extensive, it may be considered if no major risk factors or decision uncertain (Class IIb) | Extensive LGE (≥15%) is a clinical risk factor to be considered in patients in the low (to intermediate) risk category (Class IIb) | If extensive, it may be considered if no major risk factors or decision uncertain (Class IIb) |
| Genetic testing | No sufficient data | Double and compound mutations (Class IIb). Otherwise, of little prognostic value | No recommendation due to few data | No mention | Not recommended | No mention |
| Electrophysiology testing | Not recommended | Not recommended | Not recommended | No mention | No mention | No mention |
| Other | Left atrium < 45 mm compatible with lower risk | SCD risk modifiers include established risk factors and emerging risk modifiers | Class IIb recommendation for ICD when risk <4% but with features of proven prognostic significance | The HCM Risk-SCD score may help patients understand the magnitude of their risk but should not be used solely for decisions. | Clinical risk factors (Risk modifiers): LVEF < 50%, LGE (≥15%) | The HCM Risk-SCD score can be useful during the shared decision-making process (Class IIa) |
| Source | Population, n | Registry/Cohort | Genetic Focus | Main Findings |
|---|---|---|---|---|
| Watkins H et al., 1995 [71] | 27 families, 100 probands | Single center, USA | Troponin T, a-tropomyosin variants | Patients with variants in Troponin T presented with mild hypertrophy (maximum wall thickness 17 mm vs. 24 mm in patients with MYH7 variants, p < 0.001), high incidence of sudden death and a poor life expectancy of 35 years. |
| Charron P et al., 1998 [72] | 128 | Single center, France | MYBPC3 variants | Patients carrying a MYPC3 variant compared to patients carrying a MYH7 variant presented with delayed disease onset and better prognosis with no disease related deaths under the age of 40 and significantly less events of disease-related death and transplantation by 50 years of age. |
| Van Driest SL et al., 2004 [73] | 389 | Single center, USA | MYBPC3 variants | Patients with MYBPC3 variants presented with similar age at diagnosis, degree of hypertrophy, incidence of myectomy and family history of HCM or sudden death compared to patients with thick-filament HCM, thin-filament HCM or genotype-negative HCM. Patients with multiple mutations presented with the most severe disease. |
| Olivotto I et al., 2008 [74] | 203 | Two centers, Italy | Myofilament gene variants | Patients with myofilament-positive HCM showed increased risk of the combined end points of cardiovascular death, nonfatal stroke or progression to New York Heart association class III or IV compared with the patients with myofilament-negative HCM (25% vs. 7%, respectively; HR: 4.27; p = 0.008) and greater probability of severe left ventricular dysfunction. |
| Pasquale et al., 2012 [75] | 92 | Single center, UK | Troponin T variants | Among 20 probands and 72 relatives carrying TNNT2 variants, 22% received an ICD for primary prophylaxis and 4 died and the rate of the composite of cardiovascular death, transplantation and ICD discharge was 1.6% during a follow-up period of approximately 10 years. |
| Coppini et al., 2014 [76] | 230 | 4 centers in USA and Italy | Thin filament gene variants | Patients with thin-filament variants compared to those with thick-filament HCM show milder and more atypically distributed LVH, less prevalent LVOT obstruction, higher prevalence of systolic dysfunction or restricted filling and similar rates of ventricular arrhythmias and sudden cardiac death. |
| Lopes LR et al., 2015 [77] | 874 | Single center, UK | Sarcomere gene variants | Patients with sarcomere gene variants presented with younger age, more frequent family history of HCM and sudden cardiac death, greater maximum LV wall and an increased incidence of cardiovascular death (HR: 2.81, 95% CI: 1.21–6.51, p = 0.012). Similar observations were reported for individual genes. |
| Lee SP et al., 2018 [78] | 1040 | SHaRe | MYH7 variants | Patients with likely pathogenic/pathogenic MYH7 variants compared to those with MYBPC3 or thin-filament variants were younger, more likely to be probands and had the highest incidence of atrial fibrillation after adjusting for age, sex, proband status and echocardiographic parameters (hazard ratio, 1.7; 95% CI, 1.1–2.6; p = 0.009). |
| Helms A et al., 2020 [79] | 1316 | SHaRe | Truncating MYBPC3 variants | Truncating variants accounted for 91% of MYBPC3 pathogenic variants and cause similar hypertrophy independent of location. The composite endpoint (resuscitated cardiac arrest, appropriate implantable cardioverter-defibrillator therapy, heart failure outcomes, atrial fibrillation, stroke or death) did not differ among MYBPC3 variant locations and between truncating and non-truncating variants. |
| Melendo-Viu M et al., 2024 [80] | 188 | 14 centers, Spain | Truncating MYBPC3 variants | Patients with HCM carrying truncating variants presented with a low incidence of major events, most of which were heart failure. Rate of ventricular arrhythmic events was low. Neither standard risk models nor genetic factors were able to predict arrhythmic/death events. LVH showed an age-related penetrance among probands (47% at 30 years, 71% at 60 years) but the penetrance of relatives was low. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasiakogias, A.; Kaskoutis, C.; Antoniou, C.-K.; Georgopoulos, S.; Tsiachris, D.; Arsenos, P.; Kouroutzoglou, A.; Klettas, D.; Vlachopoulos, C.; Tsioufis, K.; et al. Exploring the Current Status of Risk Stratification in Hypertrophic Cardiomyopathy: From Risk Models to Promising Techniques. J. Cardiovasc. Dev. Dis. 2025, 12, 101. https://doi.org/10.3390/jcdd12030101
Kasiakogias A, Kaskoutis C, Antoniou C-K, Georgopoulos S, Tsiachris D, Arsenos P, Kouroutzoglou A, Klettas D, Vlachopoulos C, Tsioufis K, et al. Exploring the Current Status of Risk Stratification in Hypertrophic Cardiomyopathy: From Risk Models to Promising Techniques. Journal of Cardiovascular Development and Disease. 2025; 12(3):101. https://doi.org/10.3390/jcdd12030101
Chicago/Turabian StyleKasiakogias, Alexandros, Christos Kaskoutis, Christos-Konstantinos Antoniou, Stavros Georgopoulos, Dimitrios Tsiachris, Petros Arsenos, Alexandrina Kouroutzoglou, Dimitrios Klettas, Charalambos Vlachopoulos, Konstantinos Tsioufis, and et al. 2025. "Exploring the Current Status of Risk Stratification in Hypertrophic Cardiomyopathy: From Risk Models to Promising Techniques" Journal of Cardiovascular Development and Disease 12, no. 3: 101. https://doi.org/10.3390/jcdd12030101
APA StyleKasiakogias, A., Kaskoutis, C., Antoniou, C.-K., Georgopoulos, S., Tsiachris, D., Arsenos, P., Kouroutzoglou, A., Klettas, D., Vlachopoulos, C., Tsioufis, K., & Gatzoulis, K. (2025). Exploring the Current Status of Risk Stratification in Hypertrophic Cardiomyopathy: From Risk Models to Promising Techniques. Journal of Cardiovascular Development and Disease, 12(3), 101. https://doi.org/10.3390/jcdd12030101