

Lipoprotein(a): Assessing the Current Knowledge and Gaps in Screening and Treatment—A Narrative Review

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Research Strategy
2.2. Inclusion and Exclusion Criteria
2.3. Study Selection
3. Lipoprotein(a): Structure and Pathophysiology
4. Cardiovascular Risk
5. Prevention
6. Lp(a) Assessment
7. Treatment Options
7.1. Niacin
7.2. Statins
7.3. Lp(a) Apheresis
7.4. PCSK9-i
7.5. Nucleic Acid-Based Gene Silencing
7.5.1. Pelacarsen
7.5.2. Inclisiran
7.5.3. Olpasiran
7.5.4. Lepodisiran
7.5.5. Small-Molecule Inhibitors
8. Discussions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nedkoff, L.; Briffa, T.; Zemedikun, D.; Herrington, S.; Wright, F.L. Global Trends in Atherosclerotic Cardiovascular Disease. Clin. Ther. 2023, 45, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Lyngbakken, M.N.; Myhre, P.L.; Røsjø, H.; Omland, T. Novel Biomarkers of Cardiovascular Disease: Applications in Clinical Practice. Crit. Rev. Clin. Lab. Sci. 2019, 56, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Tasdighi, E.; Adhikari, R.; Almaadawy, O.; Leucker, T.M.; Blaha, M.J. LP(a): Structure, Genetics, Associated Cardiovascular Risk, and Emerging Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2024, 64, 135–157. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Malick, W.A.; Goonewardena, S.N.; Koenig, W.; Rosenson, R.S. Clinical Trial Design for Lipoprotein(a)-Lowering Therapies: JACC Focus Seminar 2/3. J. Am. Coll. Cardiol. 2023, 81, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Cho, L.; Nicholls, S.J.; Kastelein, J.; Leitersdorf, E.; Landmesser, U.; Blaha, M.; Lincoff, A.M.; Morishita, R.; et al. Lipoprotein(a) Levels in a Global Population with Established Atherosclerotic Cardiovascular Disease. Open Heart 2022, 9, e002060. [Google Scholar] [CrossRef]
- Catapano, A.L.; Tokgözoğlu, L.; Banach, M.; Gazzotti, M.; Olmastroni, E.; Casula, M.; Ray, K.K. Lipid Clinics Network Group Evaluation of Lipoprotein(a) in the Prevention and Management of Atherosclerotic Cardiovascular Disease: A Survey among the Lipid Clinics Network. Atherosclerosis 2023, 370, 5–11. [Google Scholar] [CrossRef]
- Boffa, M.B. Beyond Fibrinolysis: The Confounding Role of Lp(a) in Thrombosis. Atherosclerosis 2022, 349, 72–81. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Schneider, W.J.; Tsimikas, S.; Kovanen, P.T. Familial Hypercholesterolemia and Elevated Lipoprotein(a): Double Heritable Risk and New Therapeutic Opportunities. J. Intern. Med. 2020, 287, 2–18. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and Its Significance in Cardiovascular Disease: A Review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Vinci, P.; Di Girolamo, F.G.; Panizon, E.; Tosoni, L.M.; Cerrato, C.; Pellicori, F.; Altamura, N.; Pirulli, A.; Zaccari, M.; Biasinutto, C.; et al. Lipoprotein(a) as a Risk Factor for Cardiovascular Diseases: Pathophysiology and Treatment Perspectives. Int. J. Environ. Res. Public health 2023, 20, 6721. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F. Lipoprotein(a). In Prevention and Treatment of Atherosclerosis: Improving State-of-the-Art Management and Search for Novel Targets; von Eckardstein, A., Binder, C.J., Eds.; Springer: Cham, Switzerland, 2022; ISBN 978-3-030-86075-2. [Google Scholar]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the Kringle IV Repeat Polymorphism: The Complexity of Genetic Variation in the LPA Gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by Ethnicity and Environmental and Medical Conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef]
- Awad, K.; Mahmoud, A.K.; Abbas, M.T.; Alsidawi, S.; Ayoub, C.; Arsanjani, R.; Farina, J.M. Intra-Individual Variability in Lipoprotein(a) Levels: Findings from a Large Academic Health System Population. Eur. J. Prev. Cardiol. 2024, zwae341. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein(a) and Cardiovascular Disease. Lancet 2024, 404, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Orsó, E.; Schmitz, G. Lipoprotein(a) and Its Role in Inflammation, Atherosclerosis and Malignancies. Clin. Res. Cardiol. Suppl. 2017, 12, 31–37. [Google Scholar] [CrossRef]
- Labudovic, D.; Kostovska, I.; Tosheska Trajkovska, K.; Cekovska, S.; Brezovska Kavrakova, J.; Topuzovska, S. Lipoprotein(a)—Link between Atherogenesis and Thrombosis. Prague Med. Rep. 2019, 120, 39–51. [Google Scholar] [CrossRef]
- Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)—The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef]
- Enas, E.A.; Varkey, B.; Dharmarajan, T.S.; Pare, G.; Bahl, V.K. Lipoprotein(a): An Independent, Genetic, and Causal Factor for Cardiovascular Disease and Acute Myocardial Infarction. Indian Heart J. 2019, 71, 99–112. [Google Scholar] [CrossRef]
- Bu, L.-L.; Yuan, H.-H.; Xie, L.-L.; Guo, M.-H.; Liao, D.-F.; Zheng, X.-L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int. J. Mol. Sci. 2023, 24, 15160. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Natarajan, P.; Klarin, D.; Won, H.-H.; Peloso, G.M.; Stitziel, N.O.; Nomura, A.; Zekavat, S.M.; Bick, A.G.; et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J. Am. Coll. Cardiol. 2016, 68, 2761–2772. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-Lowering by 50 Mg/dL (105 Nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Moriarty, P.M.; Stroes, E.S. Considerations for Routinely Testing for High Lipoprotein(a). Curr. Opin. Lipidol. 2023, 34, 174. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and On-Statin Treatment Lipoprotein(a) Levels for Prediction of Cardiovascular Events: Individual Patient-Data Meta-Analysis of Statin Outcome Trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Tsimikas, S. Lipoprotein(a) and Coronary Calcium. JACC 2022, 79, 769–771. [Google Scholar] [CrossRef]
- Jackson, C.L.; Garg, P.K.; Guan, W.; Tsai, M.Y.; Criqui, M.H.; Tsimikas, S.; Bhatia, H.S. Lipoprotein(a) and Coronary Artery Calcium in Comparison with Other Lipid Biomarkers: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2023, 17, 538–548. [Google Scholar] [CrossRef]
- Bhatia, H.S. Aspirin and Lipoprotein(a) in Primary Prevention. Curr. Opin. Lipidol. 2023, 34, 214–220. [Google Scholar] [CrossRef]
- Cui, K.; Wang, H.-Y.; Yin, D.; Zhu, C.; Song, W.; Wang, H.; Jia, L.; Zhang, D.; Song, C.; Feng, L.; et al. Benefit and Risk of Prolonged Dual Antiplatelet Therapy After Percutaneous Coronary Intervention With Drug-Eluting Stents in Patients With Elevated Lipoprotein(a) Concentrations. Front. Cardiovasc. Med. 2021, 8, 807925. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Koschinsky, M.L.; Albers, J.J.; Skarlatos, S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: Recent Advances and Future Directions. Clin. Chem. 2003, 49, 1785–1796. [Google Scholar] [CrossRef]
- Minelli, S.; Minelli, P.; Montinari, M.R. Reflections on Atherosclerosis: Lesson from the Past and Future Research Directions. J. Multidiscip. Healthc. 2020, 13, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, S.; Tan, J.; Wei, L.; Wu, D.; Gao, S.; Weng, Y.; Chen, J. Recent Advance in Treatment of Atherosclerosis: Key Targets and Plaque-Positioned Delivery Strategies. J. Tissue Eng. 2022, 13, 20417314221088509. [Google Scholar] [CrossRef]
- Pirillo, A.; Bonacina, F.; Norata, G.D.; Catapano, A.L. The Interplay of Lipids, Lipoproteins, and Immunity in Atherosclerosis. Curr. Atheroscler. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Scanu, A.M.; Kennedy, H.; Giaculli, F.; Berg, K.; Couderc, R.; Dati, F.; Rifai, N.; Sakurabayashi, I.; et al. Use of a Reference Material Proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to Evaluate Analytical Methods for the Determination of Plasma Lipoprotein(a). Clin. Chem. 2000, 46, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- Borque, L.; Rus, A.; del Cura, J.; Maside, C.; Escanero, J. Automated Latex Nephelometric Immunoassay for the Measurement of Serum Lipoprotein (a). J. Clin. Lab. Anal. 1993, 7, 105–110. [Google Scholar] [CrossRef]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and How to Measure It. Ann. Clin. Biochem. 2021, 58, 16–21. [Google Scholar] [CrossRef]
- Heydari, M.; Rezayi, M.; Ruscica, M.; Jpamialahamdi, T.; Johnston, T.P.; Sahebkar, A. The Ins and Outs of Lipoprotein(a) Assay Methods. Arch. Med. Sci. Atheroscler. Dis. 2023, 8, e128–e139. [Google Scholar] [CrossRef]
- Szarek, M.; Reijnders, E.; Jukema, J.W.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Fazio, S.; Garon, G.; Goodman, S.G.; Harrington, R.A.; et al. Relating Lipoprotein(a) Concentrations to Cardiovascular Event Risk After Acute Coronary Syndrome: A Comparison of 3 Tests. Circulation 2024, 149, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Shahjehan, R.D.; Sharma, S.; Bhutta, B.S. Coronary Artery Disease; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F. The Promise of PCSK9 and Lipoprotein(a) as Targets for Gene Silencing Therapies. Clin. Ther. 2023, 45, 1034–1046. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendia, L.E.; Ferretti, G.; Cicero, A.F. Effect of Extended-Release Niacin on Plasma Lipoprotein(a) Levels: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. Metab. Clin. Exp. 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Ballantyne, C.M. Existing and Emerging Strategies to Lower Lipoprotein(a). Atherosclerosis 2022, 349, 110–122. [Google Scholar] [CrossRef]
- Tsaban, G. Statins and Lipoprotein(a); Facing the Residual Risk. Eur. J. Prev. Cardiol. 2022, 29, 777–778. [Google Scholar] [CrossRef]
- de Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin Therapy and Lipoprotein(a) Levels: A Systematic Review and Meta-Analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Safarova, M.S.; Moriarty, P.M. Lipoprotein Apheresis: Current Recommendations for Treating Familial Hypercholesterolemia and Elevated Lipoprotein(a). Curr. Atheroscler. Rep. 2023, 25, 391–404. [Google Scholar] [CrossRef]
- Víšek, J.; Bláha, M.; Bláha, V.; Lášticová, M.; Lánska, M.; Andrýs, C.; Tebbens, J.D.; Igreja e Sá, I.C.; Tripská, K.; Vicen, M.; et al. Monitoring of up to 15 Years Effects of Lipoprotein Apheresis on Lipids, Biomarkers of Inflammation, and Soluble Endoglin in Familial Hypercholesterolemia Patients. Orphanet. J. Rare Dis. 2021, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Schettler, V.J.; Peter, C.; Zimmermann, T.; Julius, U.; Roeseler, E.; Schlieper, G.; Heigl, F.; Grützmacher, P.; Löhlein, I.; Klingel, R.; et al. The German Lipoprotein Apheresis Registry—Summary of the Ninth Annual Report. Ther. Apher. Dial. 2022, 26, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R. The Scientific Basis and Future of Lipoprotein Apheresis. Ther. Apher. Dial. 2022, 26, 32–36. [Google Scholar] [CrossRef]
- Nugent, A.K.; Gray, J.V.; Gorby, L.K.; Moriarty, P.M. Lipoprotein Apheresis: First FDA Indicated Treatment for Elevated Lipoprotein(a). J. Clin. Cardiol. 2020, 1, 16–21. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Karsten, V.; Chan, A.; Chiesa, J.; Boyce, M.; Bettencourt, B.R.; Hutabarat, R.; Nochur, S.; Vaishnaw, A.; Gollob, J. Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol. Ther. 2017, 25, 71–78. [Google Scholar] [CrossRef]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A New Dawn for Managing Dyslipidemias: The Era of Rna-Based Therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef]
- Landmesser, U.; Poller, W.; Tsimikas, S.; Most, P.; Paneni, F.; Lüscher, T.F. From Traditional Pharmacological towards Nucleic Acid-Based Therapies for Cardiovascular Diseases. Eur. Heart J. 2020, 41, 3884–3899. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Packard, C.J.; Chapman, M.J.; Katzmann, I.; Laufs, U. Targeting RNA With Antisense Oligonucleotides and Small Interfering RNA: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 563–579. [Google Scholar] [CrossRef]
- Yeang, C.; Karwatowska-Prokopczuk, E.; Su, F.; Dinh, B.; Xia, S.; Witztum, J.L.; Tsimikas, S. Effect of Pelacarsen on Lipoprotein(a) Cholesterol and Corrected Low-Density Lipoprotein Cholesterol. J. Am. Coll. Cardiol. 2022, 79, 1035–1046. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. A Randomized Double-Blind, Placebo-Controlled, Multicenter Trial Assessing the Impact of Lipoprotein (a) Lowering with Pelacarsen (TQJ230) on Major Cardiovascular Events in Patients with Established Cardiovascular Disease. 2025. Available online: https://clinicaltrials.gov/study/NCT04023552 (accessed on 3 February 2025).
- Katsiki, N.; Vrablik, M.; Banach, M.; Gouni-Berthold, I. Inclisiran, Low-Density Lipoprotein Cholesterol and Lipoprotein (a). Pharmaceuticals 2023, 16, 577. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Eli Lilly and Company. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Investigate the Efficacy and Safety of LY3819469 in Adults with Elevated Lipoprotein(a). 2025. Available online: https://clinicaltrials.gov/study/NCT05565742 (accessed on 3 February 2025).
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.F.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an Extended-Duration Short Interfering RNA Targeting Lipoprotein(a). JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Hooper, A.J.; Fernando, P.M.S.; Burnett, J.R. Potential of Muvalaplin as a Lipoprotein(a) Inhibitor. Expert Opin. Investig. Drugs 2024, 33, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ni, W.; Rhodes, G.M.; Nissen, S.E.; Navar, A.M.; Michael, L.F.; Haupt, A.; Krege, J.H. Oral Muvalaplin for Lowering of Lipoprotein(a): A Randomized Clinical Trial. JAMA 2025, 333, 222–231. [Google Scholar] [CrossRef]
- Chakraborty, A.; Pang, J.; Chan, D.C.; Ellis, K.L.; Hooper, A.J.; Bell, D.A.; Burnett, J.R.; Moses, E.K.; Watts, G.F. Cascade Testing for Elevated Lipoprotein(a) in Relatives of Probands with Familial Hypercholesterolaemia and Elevated Lipoprotein(a). Atherosclerosis 2022, 349, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Annink, M.E.; Janssen, E.S.; Reeskamp, L.F. Effectiveness of Cascade Screening for Elevated Lipoprotein(a), an Underdiagnosed Family Disorder. Curr. Opin. Lipidol. 2024, 35, 290. [Google Scholar] [CrossRef]
- De Vries, T.I.; Cooney, M.T.; Selmer, R.M.; Hageman, S.H.; Pennells, L.A.; Wood, A.; Kaptoge, S.; Xu, Z.; Westerink, J.; Rabanal, K.S.; et al. SCORE2-OP Risk Prediction Algorithms: Estimating Incident Cardiovascular Event Risk in Older Persons in Four Geographical Risk Regions. Eur. Heart J. 2021, 42, 2455–2467. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Assesment Method | Description | Advantages | Limitations | Cut-Off Value |
|---|---|---|---|---|
| Immunoturbidimetric Assay | Measures turbidity changes due to Lp(a)–antibody complexes | Broad range; High-throughput | Influenced by apo(a) size; Variability between reagents | LOW |
| Nephelometry | Measures light scattering from antigen–antibody complexes | Automated; Reproducible; Broad detection range | Influenced by apo(a) size; Less standardization | <30 mg/dL or <75 nmol/L |
| ELISA | Uses antibodies targeting Lp(a) components | Sensitive; Adaptable | Accuracy depends on antibody used | INCREASED RISK |
| Denka Assay | Commercial assay with multiple calibrators for isoform coverage | Least affected by isoform heterogeneity; More accurate and standardized | Dependent on proper calibration; Available through specific providers | 30–50 mg/dL or 75–125 nmol/L |
| Radial Immunodiffusion | Simple diffusion-based immunoassay measuring precipitate ring | Low cost; Detects low levels of Lp(a) | Low sensitivity; Slow; Cannot assess apo(a) isoform size | HIGH >50 mg/dL or >125 nmol/L |
| Mass Spectrometry | Direct quantification of apolipoprotein(a) peptides | Isoform-independent; Accurate; Better standardization | Expensive; Not widely available; Complex technique |
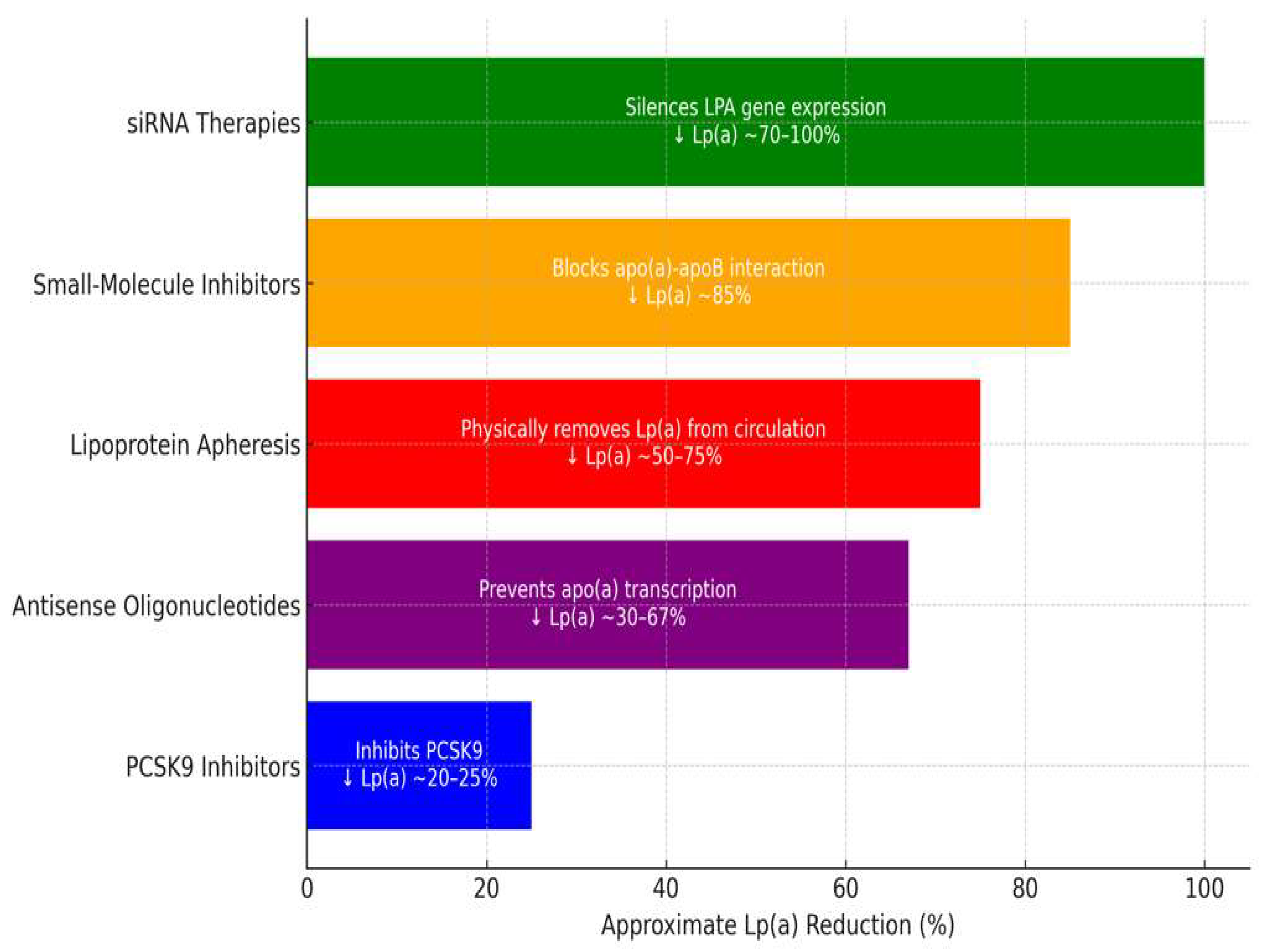
| Therapy | Mechanism of Action | Reduction in Lp(a) | Duration of Effect | Limitations |
|---|---|---|---|---|
| PCSK9-i | Increases clearance via LDLR upregulation | 20–25% | Short-term, frequent dosing | Moderate effect on Lp(a) reduction |
| ASOs (Pelacarsen) | Inhibits apo(a) mRNA translation | 29–67% | Long-term, monthly dosing | Not yet widely available |
| siRNA (Olpasiran) | Blocks apo(a) mRNA translation | 68.5–100% | Long-term, quarterly dosing | Not yet widely available |
| Apheresis | Physically removes Lp(a) from circulation | up to 73% | Immediate but transient | Invasive, expensive |
| Small-molecule inhibitors (Muvalaplin) | Disrupts Lp(a) assembly | up to 85.8% | Oral, daily dosing | Early-stage development |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaritei, O.; Mierlan, O.L.; Gutu, C.; Gurau, G. Lipoprotein(a): Assessing the Current Knowledge and Gaps in Screening and Treatment—A Narrative Review. J. Cardiovasc. Dev. Dis. 2025, 12, 169. https://doi.org/10.3390/jcdd12050169
Amaritei O, Mierlan OL, Gutu C, Gurau G. Lipoprotein(a): Assessing the Current Knowledge and Gaps in Screening and Treatment—A Narrative Review. Journal of Cardiovascular Development and Disease. 2025; 12(5):169. https://doi.org/10.3390/jcdd12050169
Chicago/Turabian StyleAmaritei, Octavian, Oana Laura Mierlan, Cristian Gutu, and Gabriela Gurau. 2025. "Lipoprotein(a): Assessing the Current Knowledge and Gaps in Screening and Treatment—A Narrative Review" Journal of Cardiovascular Development and Disease 12, no. 5: 169. https://doi.org/10.3390/jcdd12050169
APA StyleAmaritei, O., Mierlan, O. L., Gutu, C., & Gurau, G. (2025). Lipoprotein(a): Assessing the Current Knowledge and Gaps in Screening and Treatment—A Narrative Review. Journal of Cardiovascular Development and Disease, 12(5), 169. https://doi.org/10.3390/jcdd12050169