
Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background

Abstract
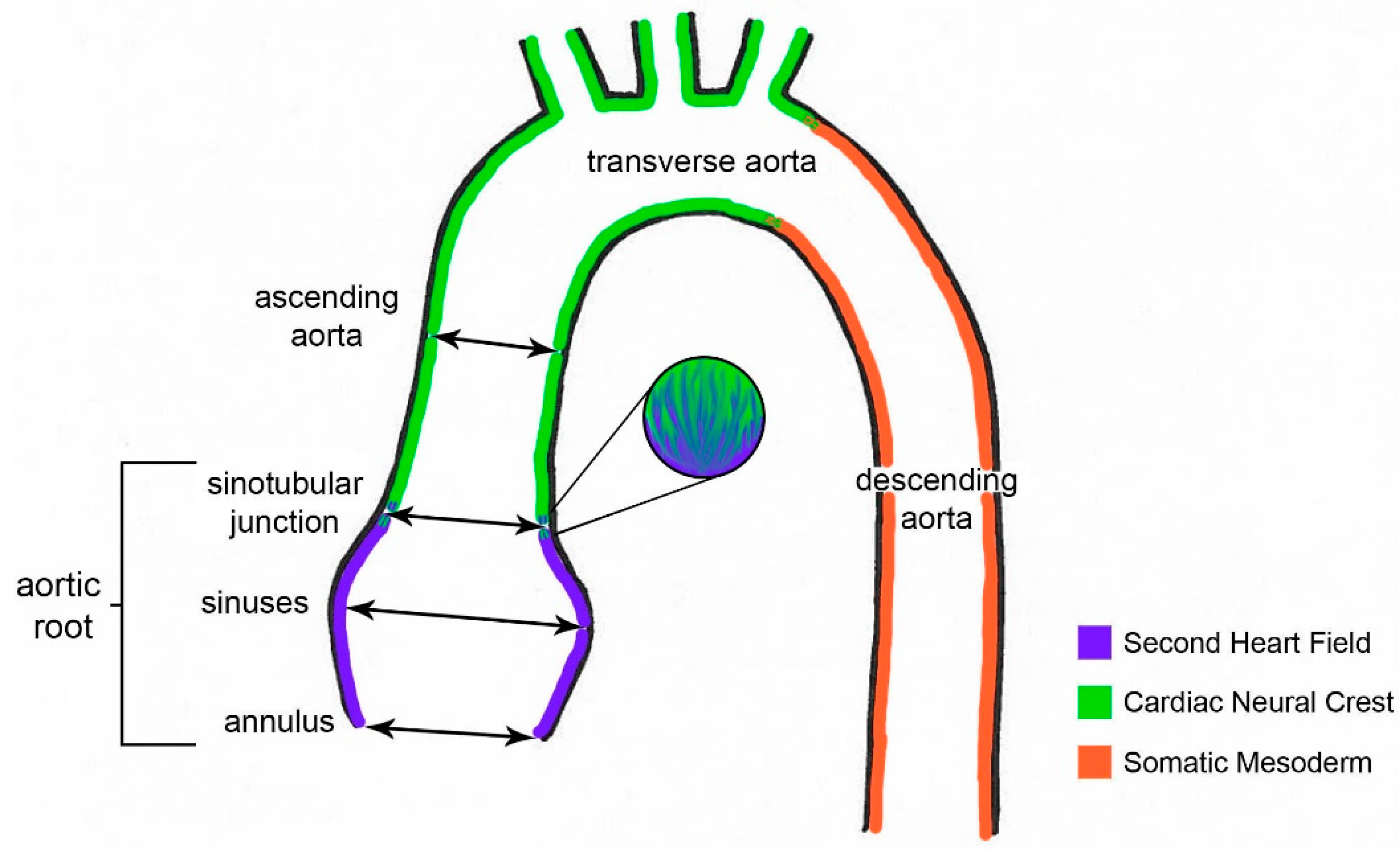
:1. Introduction

2. Experimental Section
2.1. Mice
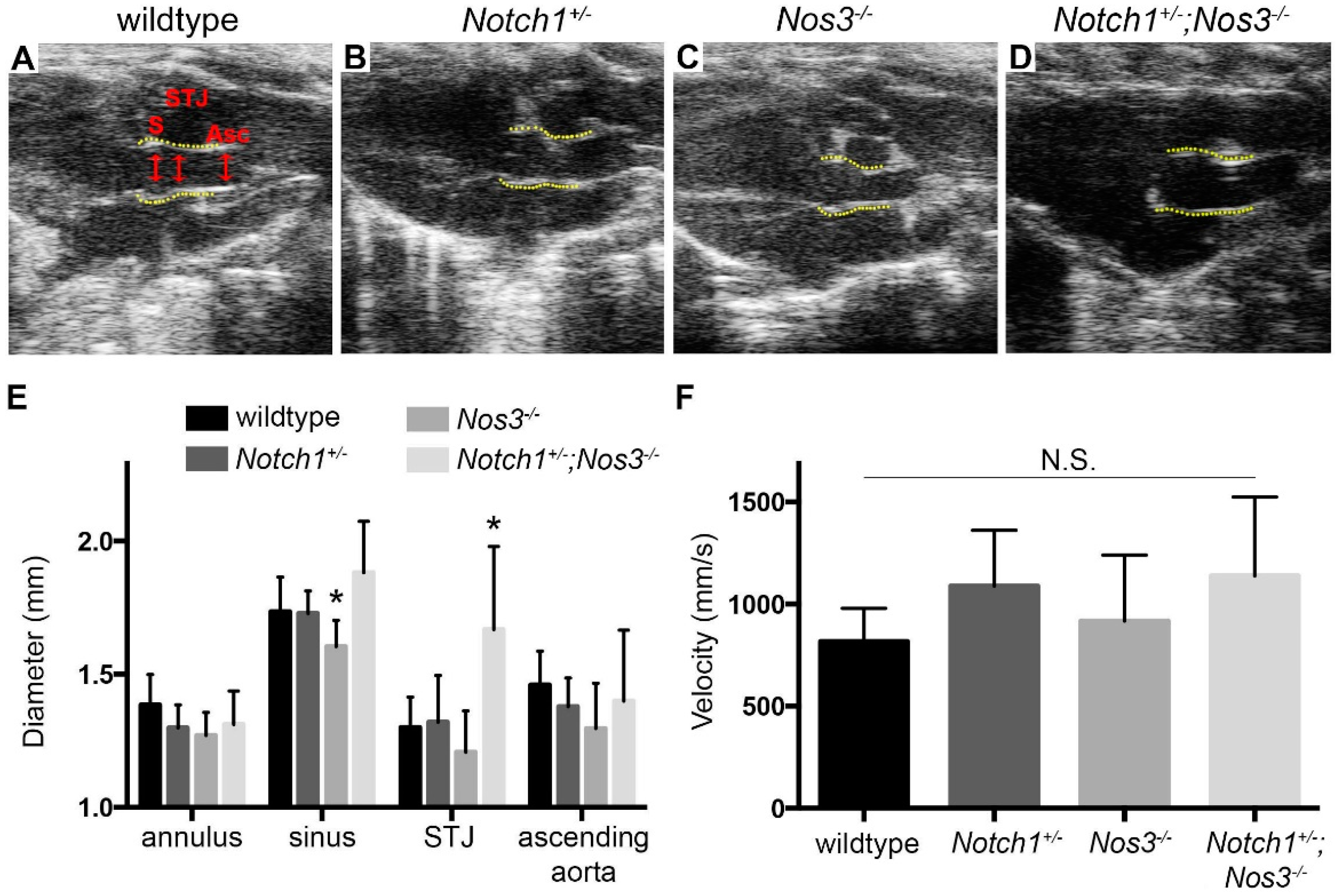
2.2. Echocardiography

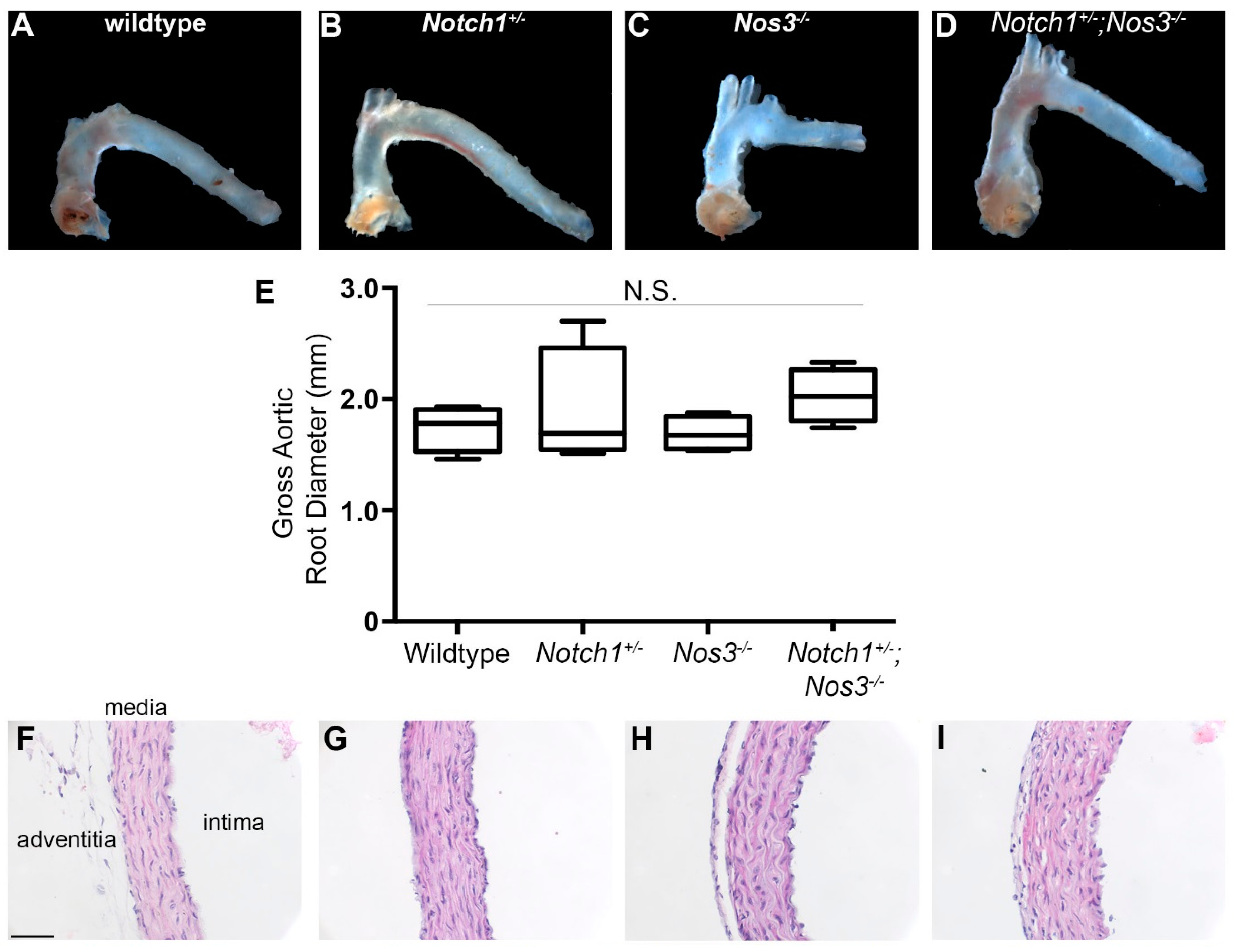
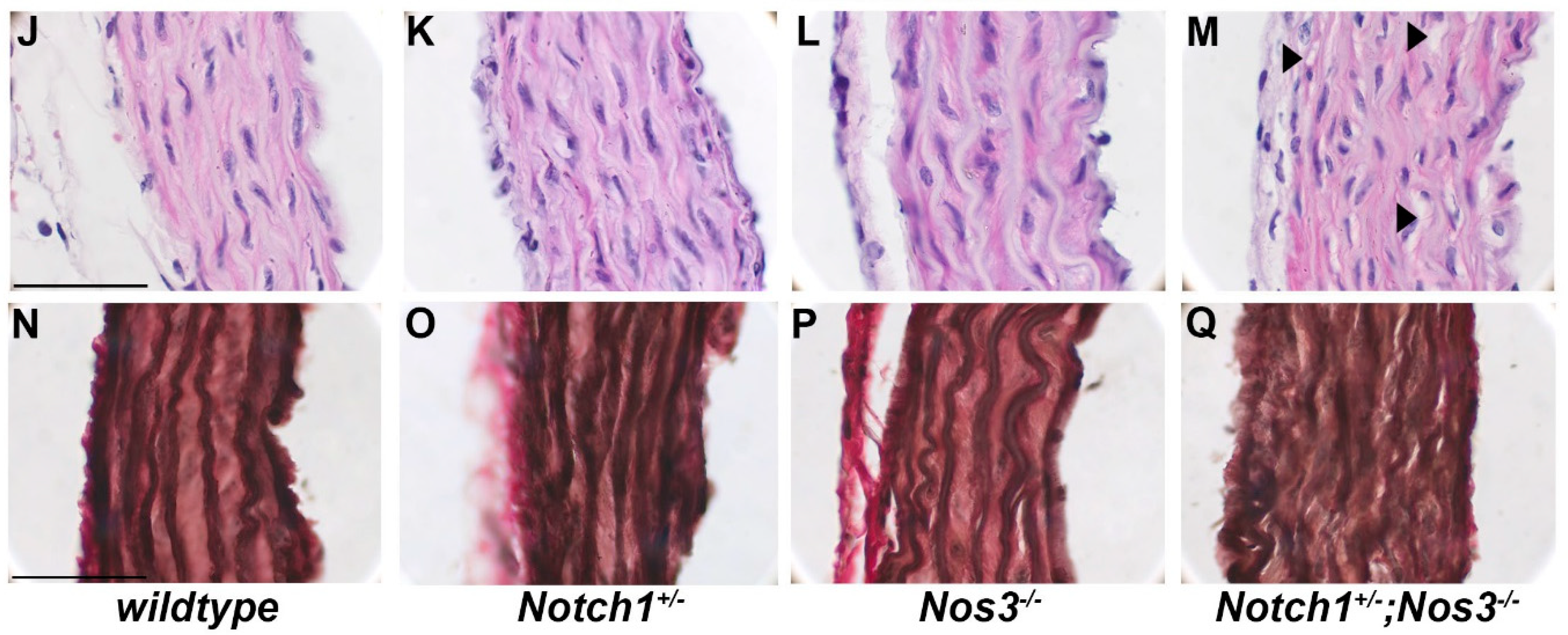
2.3. Tissue Fixation and Histology
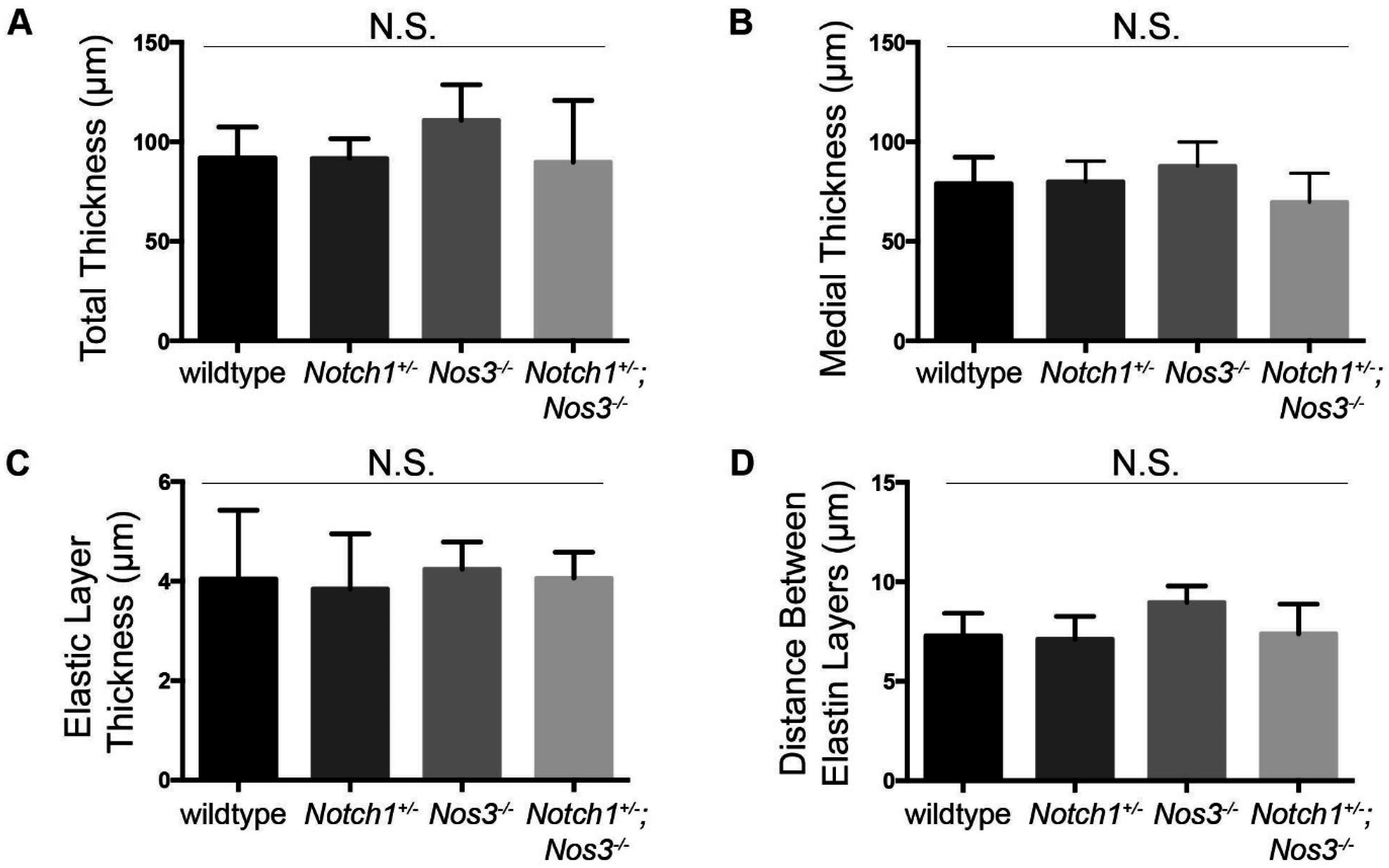
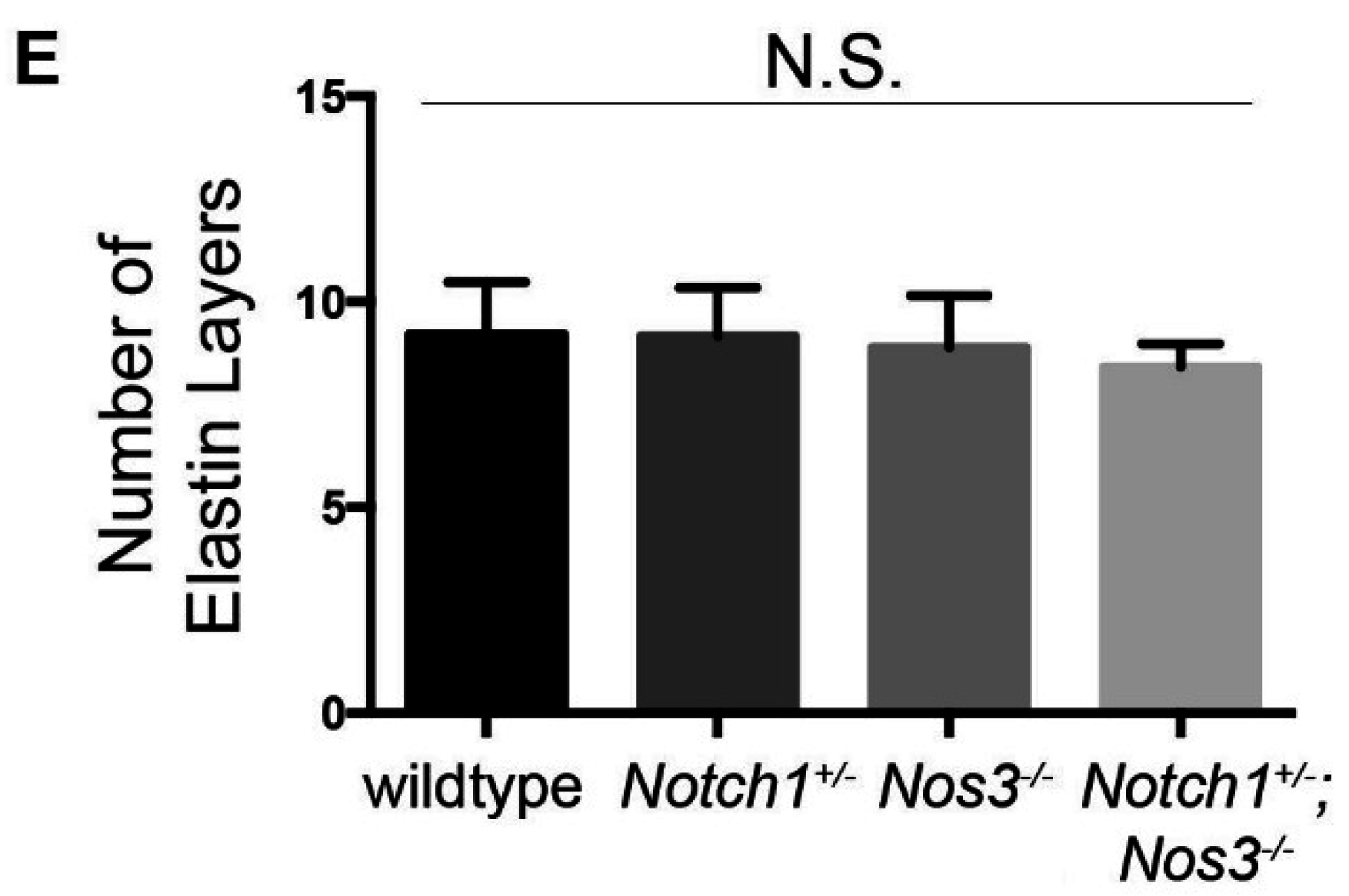
2.4. Elastin Quantification and Basic Morphometrics
2.5. TGF-β1 ELISA
2.6. Statistics
3. Results and Discussion
3.1. Effacement of Sinotubular Junction and Aortic Root Dilation in Notch1+/−; Nos3−/− Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wildtype | Notch1+/− | Nos3−/− | Notch1+/−; Nos3−/− | |
|---|---|---|---|---|
| 3 months | 0/8 (0%) | 1/8 (12.5%) | 3/11 (27.3%) | 5/5 (100%) * |
| 6 months | 0/8 (0%) | 1/8 (12.5%) | 4/11 (36.4%) | 5/5 (100%) * |
3.2. Notch1+/−; Nos3−/− Mice Display Gross and Histologic Evidence of Aortopathy


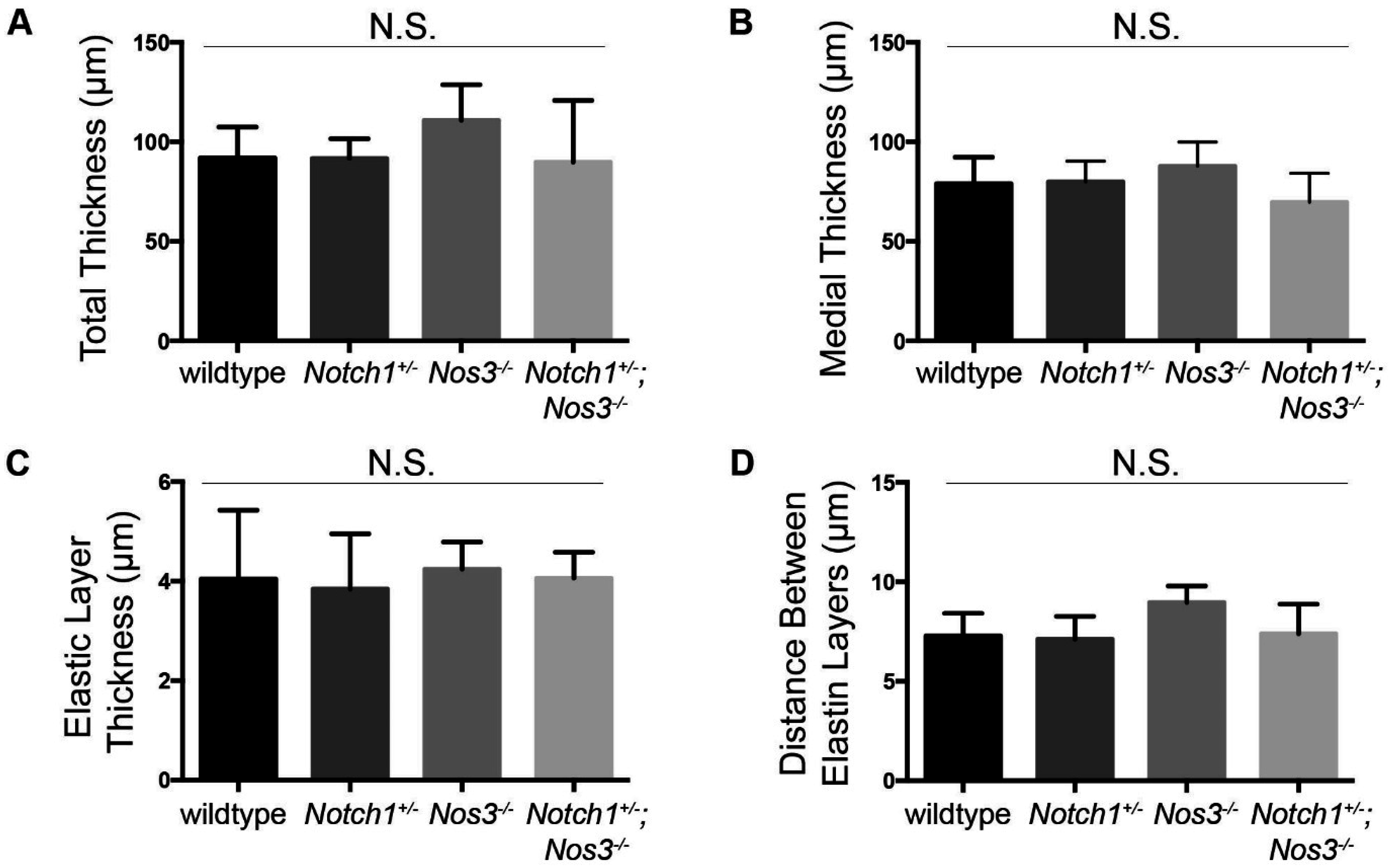

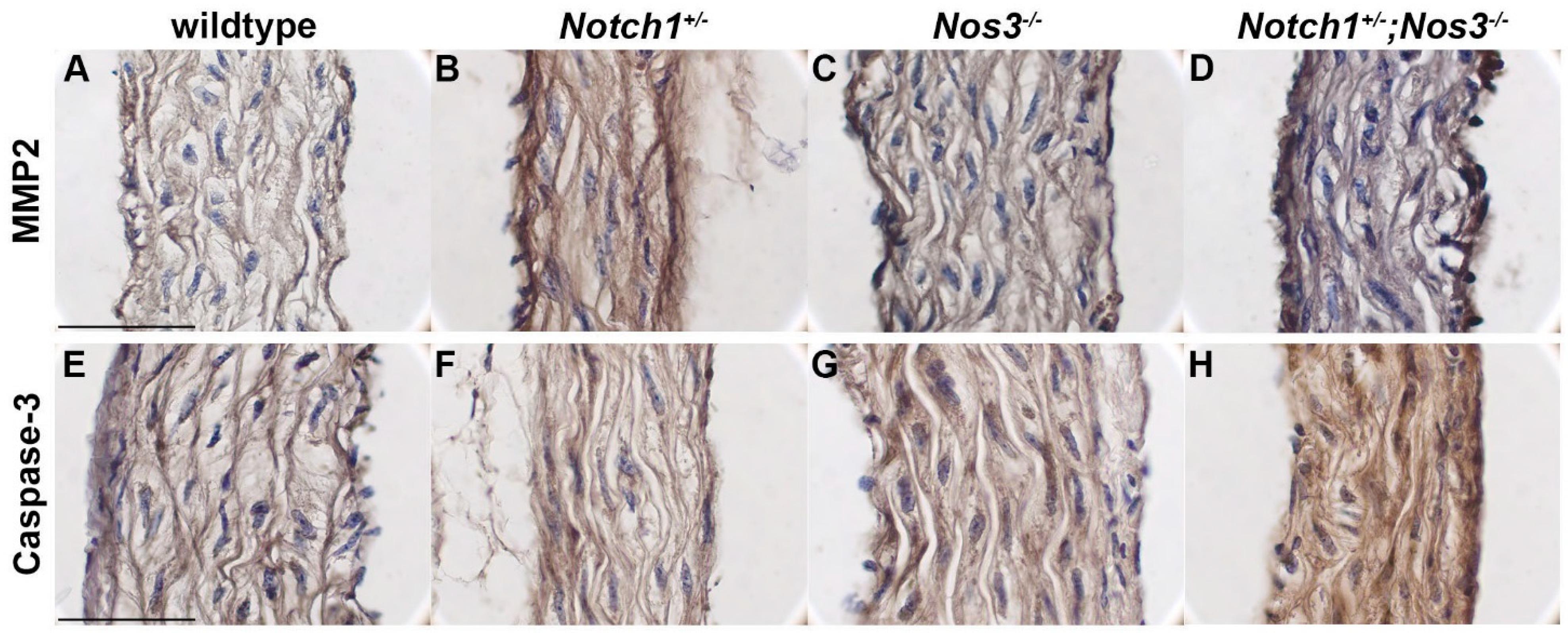
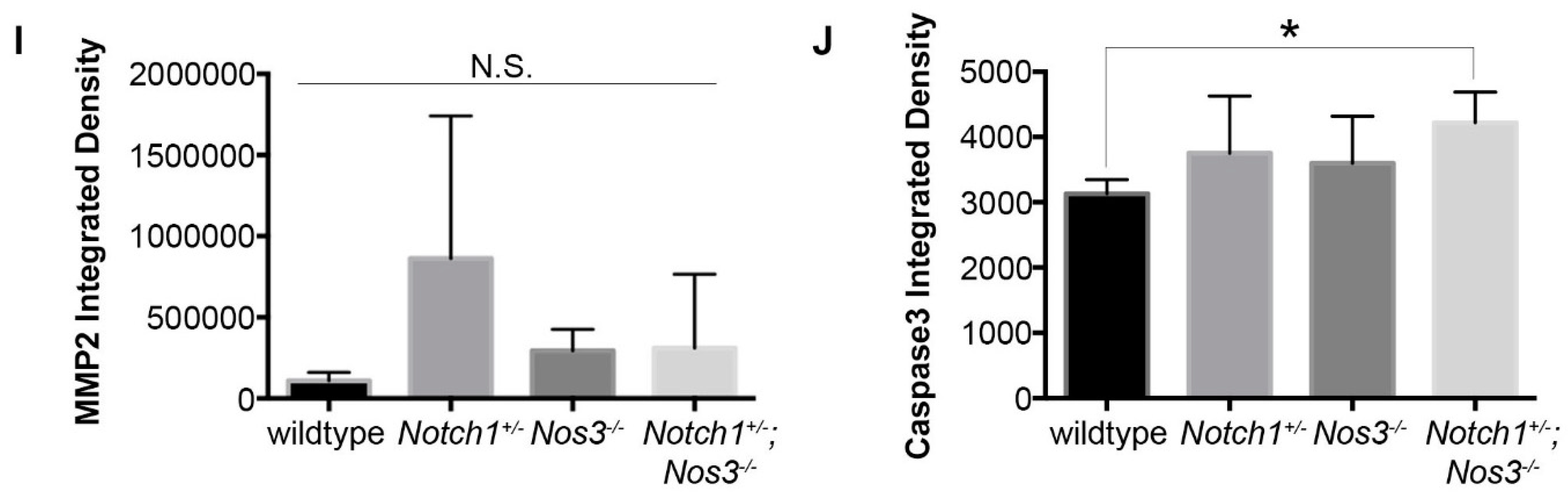
3.3. Notch1+/−; Nos3−/− Aortas Display Molecular Evidence of Aortopathy


3.4. Significance
4. Limitations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clouse, W.D.; Hallett, J.W., Jr.; Schaff, H.V.; Spittell, P.C.; Rowland, C.M.; Ilstrup, D.M.; Melton, L.J., 3rd. Acute aortic dissection: Population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin. Proc. 2004, 79, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Paterick, T.E.; Humphries, J.A.; Ammar, K.A.; Jan, M.F.; Loberg, R.; Bush, M.; Khandheria, B.K.; Tajik, A.J. Aortopathies: Etiologies, genetics, differential diagnosis, prognosis and management. Am. J. Med. 2013, 126, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, F.; Mazzaglia, D.; Pellegrino, A.; Grego, S.; Fiorito, R.; Ferlosio, A.; Chiariello, L.; Orlandi, A. Aortopathy in marfan syndrome: An update. Cardiovasc. Pathol. 2014, 23, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, K.; Pulkkinen, L.; Savolainen, A.; Kaitila, I.; Peltonen, L. Location on chromosome 15 of the gene defect causing marfan syndrome. N. Engl. J. Med. 1990, 323, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Godfrey, M.; Vitale, E.; Hori, H.; Mattei, M.G.; Sarfarazi, M.; Tsipouras, P.; Ramirez, F.; Hollister, D.W. Linkage of marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature 1991, 352, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Maslen, C.L.; Corson, G.M.; Maddox, B.K.; Glanville, R.W.; Sakai, L.Y. Partial sequence of a candidate gene for the marfan syndrome. Nature 1991, 352, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Vranckx, R.; Khau van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palacek, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in fabry disease. Eur. Heart J. 2010, 31, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of ehlers-danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; de Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the tgf-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Gurvitz, M.; Chang, R.K.; Drant, S.; Allada, V. Frequency of aortic root dilation in children with a bicuspid aortic valve. Am. J. Cardiol. 2004, 94, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, M.; Manfrin, M.; Moraca, A.; Zanoli, R.; Colonna, P.L.; Bettuzzi, M.G.; Moretti, S.; Gabrielli, D.; Perna, G.P. Aortic dimensions in patients with bicuspid aortic valve without significant valve dysfunction. Am. J. Cardiol. 2005, 95, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Siu, S.C. Aortic dilatation in patients with bicuspid aortic valve. N. Engl. J. Med. 2014, 370, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; Sorbo, M.D.; Marin, M.; Palisi, M.; Scognamiglio, R.; Thiene, G. Aortic root dilatation in young men with normally functioning bicuspid aortic valves. Heart 1999, 82, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Arheart, K.L.; Colan, S.D.; Stein, N.S.; Lopez-Mitnik, G.; Lin, A.E.; Reller, M.D.; Ventura, R.; Silberbach, M. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics 2008, 121, e1622–e1627. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.H.; Hjerrild, B.E.; Stochholm, K.; Andersen, N.H.; Sorensen, K.E.; Lundorf, E.; Horlyck, A.; Pedersen, E.M.; Christiansen, J.S.; Gravholt, C.H. Dilation of the ascending aorta in turner syndrome—A prospective cardiovascular magnetic resonance study. J. Cardiov. Magn. Reson. 2011, 13, 24. [Google Scholar] [CrossRef]
- Olivieri, L.J.; Baba, R.Y.; Arai, A.E.; Bandettini, W.P.; Rosing, D.R.; Bakalov, V.; Sachdev, V.; Bondy, C.A. Spectrum of aortic valve abnormalities associated with aortic dilation across age groups in turner syndrome. Circul. Cardiovasc. Imaging 2013, 6, 1018–1023. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in Notch1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- De la Pompa, J.L.; Epstein, J.A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev. Cell 2012, 22, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [PubMed]
- Jain, R.; Rentschler, S.; Epstein, J.A. Notch and cardiac outflow tract development. Ann. N. Y. Acad. Sci. 2010, 1188, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [PubMed]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M., 3rd. Novel Notch1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.; Dietz, H.C. Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146. [Google Scholar] [CrossRef]
- Sciacca, S.; Pilato, M.; Mazzoccoli, G.; Pazienza, V.; Vinciguerra, M. Anti-correlation between longevity gene SirT1 and Notch signaling in ascending aorta biopsies from patients with bicuspid aortic valve disease. Heart Vessels 2013, 28, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Ren, P.; Nguyen, M.; Coselli, J.S.; Shen, Y.H.; LeMaire, S.A. Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS One 2012, 7, e52833. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell. Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef] [PubMed]
- Verzi, M.P.; McCulley, D.J.; de Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyurek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Qiu, G.; Ilyas, M. Immunohistochemistry (IHC) image analysis toolbox. Available online: http://rsb.info.nih.gov/ij/plugins/ihc-toolbox/index.html (accessed on 4 March 2015).
- Koenig, S.N.; Garg, V. Aortic diameter measurements of 3 month old Notch1+/−; Nos3−/− mice. Nationwide Children’s Hospital: Columbus, OH, USA, Unpublished data. 2014. [Google Scholar]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending aortic dilatation associated with bicuspid aortic valve: Pathophysiology, molecular biology, and clinical implications. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Kennedy, M.; Kazarov, A.; Papadimitriou, J.C.; Keller, G. A common precursor for hematopoietic and endothelial cells. Development 1998, 125, 725–732. [Google Scholar] [PubMed]
- Takeshita, K.; Satoh, M.; Ii, M.; Silver, M.; Limbourg, F.P.; Mukai, Y.; Rikitake, Y.; Radtke, F.; Gridley, T.; Losordo, D.W.; et al. Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ. Res. 2007, 100, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Hope, T.A.; Markl, M.; Wigstrom, L.; Alley, M.T.; Miller, D.C.; Herfkens, R.J. Comparison of flow patterns in ascending aortic aneurysms and volunteers using four-dimensional magnetic resonance velocity mapping. JMRI 2007, 26, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Chien, S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1209–H1224. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, M.; Millot, N.; Sheikhzadeh, S.; Rybczynski, M.; Gerth, S.; Kolbel, T.; Keyser, B.; Kutsche, K.; Robinson, P.N.; Berger, J.; et al. Total serum transforming growth factor-beta1 is elevated in the entire spectrum of genetic aortic syndromes. Clin. Cardiol. 2014, 37, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.; van Laer, L.; Loeys, B.L. Genetics of thoracic aortic aneurysm: At the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ. Res. 2013, 113, 327–340. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koenig, S.N.; Bosse, K.M.; Nadorlik, H.A.; Lilly, B.; Garg, V. Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background. J. Cardiovasc. Dev. Dis. 2015, 2, 17-30. https://doi.org/10.3390/jcdd2010017
Koenig SN, Bosse KM, Nadorlik HA, Lilly B, Garg V. Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background. Journal of Cardiovascular Development and Disease. 2015; 2(1):17-30. https://doi.org/10.3390/jcdd2010017
Chicago/Turabian StyleKoenig, Sara N., Kevin M. Bosse, Holly A. Nadorlik, Brenda Lilly, and Vidu Garg. 2015. "Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background" Journal of Cardiovascular Development and Disease 2, no. 1: 17-30. https://doi.org/10.3390/jcdd2010017
APA StyleKoenig, S. N., Bosse, K. M., Nadorlik, H. A., Lilly, B., & Garg, V. (2015). Evidence of Aortopathy in Mice with Haploinsufficiency of Notch1 in Nos3-Null Background. Journal of Cardiovascular Development and Disease, 2(1), 17-30. https://doi.org/10.3390/jcdd2010017