Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ApoE and Plasma Lipid Homeostasis
3. ApoE4 Domain Interaction and Atherosclerosis
4. Pleiotropic Properties of ApoE in Inflammation and Atherosclerosis Control
5. ApoE Regulation of Macrophage Polarity and Inflammatory Phenotypes
6. Cell-Derived ApoE and Control of Lipid-Induced HSPC Proliferation
7. Plasma ApoE and Control of Monocyte Activation in Hyperlipidemic Mice
8. ApoE Remodels HDL and Improves their Function in Hyperlipidemia
9. ApoE Regulation of Myeloid Cells Signaling via MicroRNA: Impact on Atherosclerosis
10. Conclusions
Funding
Conflicts of Interest
References
- Davignon, J. Apolipoprotein e and atherosclerosis: Beyond lipid effect. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K. Apoe in atherosclerosis. A protein with multiple hats. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1852–1853. [Google Scholar] [CrossRef] [PubMed]
- Raffai, R.L. Apolipoprotein E regulation of myeloid cell plasticity in atherosclerosis. Curr. Opin. Lipidol. 2012, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Fazio, S.; Babaev, V.R.; Murray, A.B.; Hasty, A.H.; Carter, K.J.; Gleaves, L.A.; Atkinson, J.B.; Linton, M.F. Increased atherosclerosis in mice reconstituted with apolipoprotein e null macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 4647–4652. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Boisvert, W.A. Apolipoprotein e and atherosclerosis. Curr. Opin. Lipidol. 2000, 11, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. Apoe regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
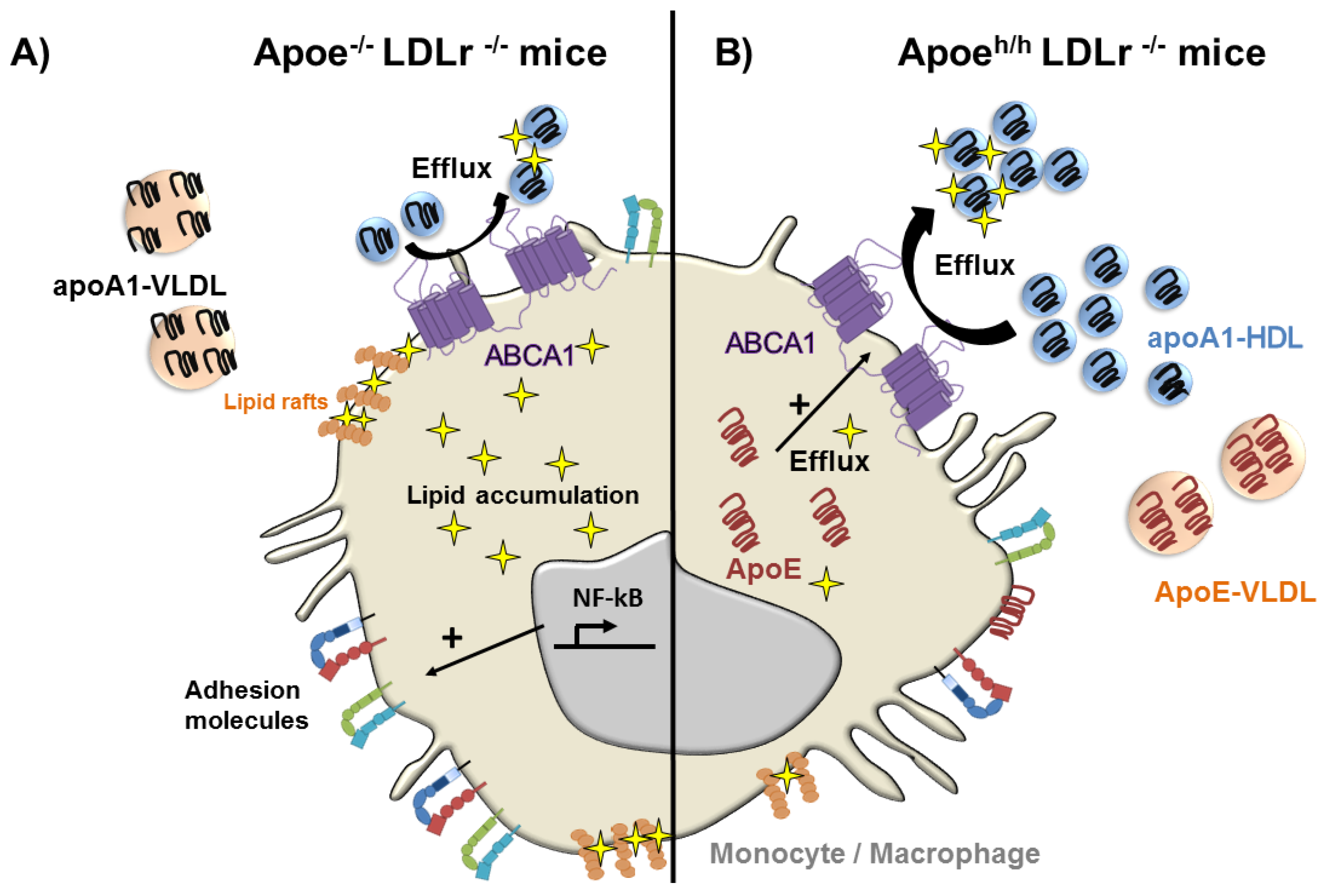
- Gaudreault, N.; Kumar, N.; Posada, J.M.; Stephens, K.B.; Reyes de Mochel, N.S.; Eberle, D.; Olivas, V.R.; Kim, R.Y.; Harms, M.J.; Johnson, S.; et al. Apoe suppresses atherosclerosis by reducing lipid accumulation in circulating monocytes and the expression of inflammatory molecules on monocytes and vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, D.; Castagnino, P.; Rader, D.J.; Phillips, M.C.; Lund-Katz, S.; Assoian, R.K. Apolipoprotein e-mediated cell cycle arrest linked to p27 and the cox2-dependent repression of mir221/222. Atherosclerosis 2013, 227, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by Apoe and Apoe-HDL linked to suppression of ecm gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
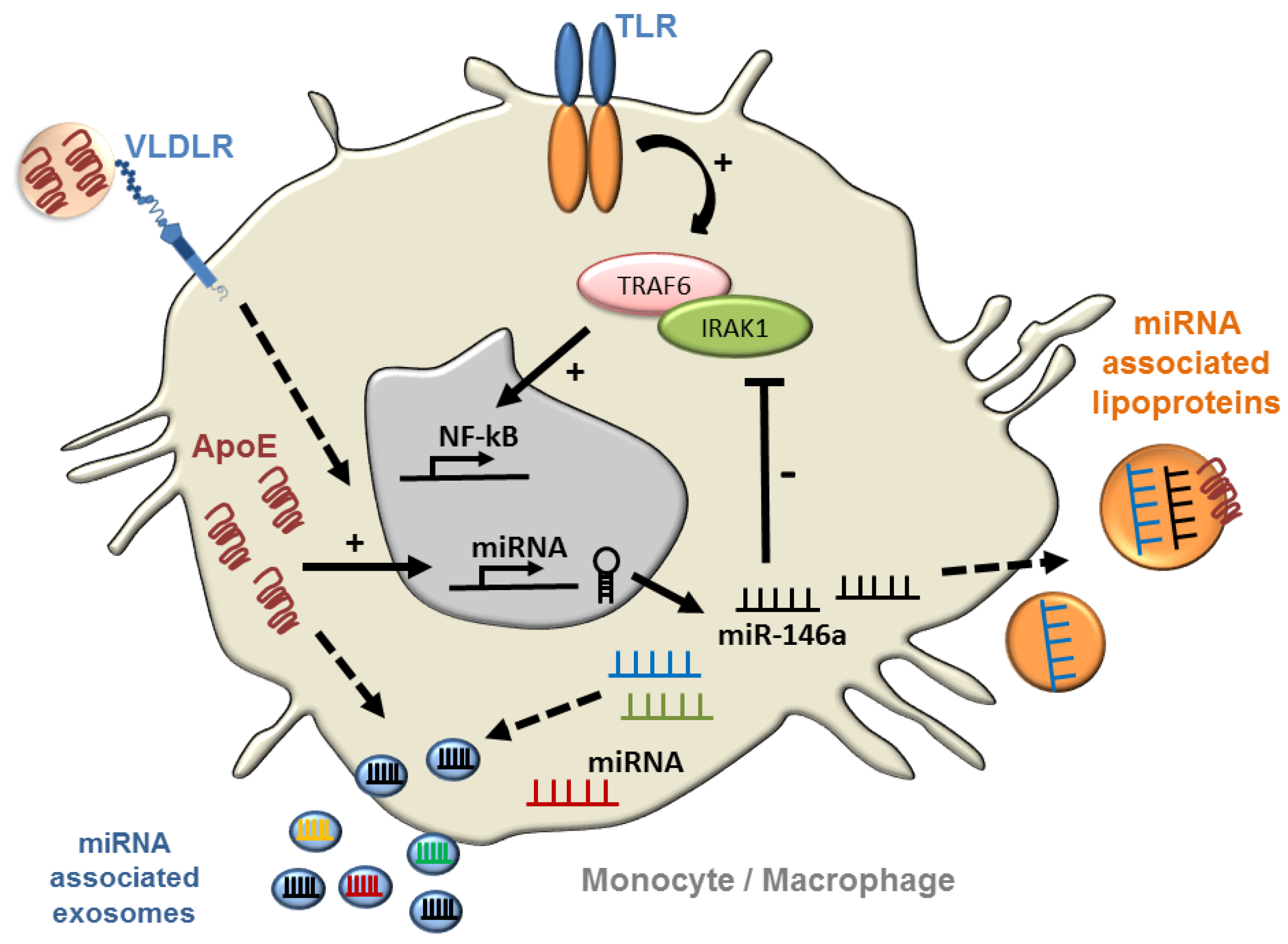
- Li, K.; Ching, D.; Luk, F.S.; Raffai, R.L. Apolipoprotein e enhances microRNA-146a in monocytes and macrophages to suppress nuclear factor-kappab-driven inflammation and atherosclerosis. Circ. Res. 2015, 117, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein e: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Mahley, R.W.; Boyles, J.K.; Fainaru, M.; Brecht, W.J.; Lindquist, P.A. Chylomicron-chylomicron remnant clearance by liver and bone marrow in rabbits. Factors that modify tissue-specific uptake. J. Biol. Chem. 1989, 264, 9571–9582. [Google Scholar] [PubMed]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C., Jr.; Weisgraber, K.H.; Taylor, J.M. Apolipoprotein E: Genetic variants provide insights into its structure and function. Curr. Opin. Lipidol. 1990, 1, 87–95. [Google Scholar] [CrossRef]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein e. J. Lipid Res. 1999, 40, 1–16. [Google Scholar] [PubMed]
- Hasty, A.H.; Linton, M.F.; Swift, L.L.; Fazio, S. Determination of the lower threshold of apolipoprotein e resulting in remnant lipoprotein clearance. J. Lipid Res. 1999, 40, 1529–1538. [Google Scholar] [PubMed]
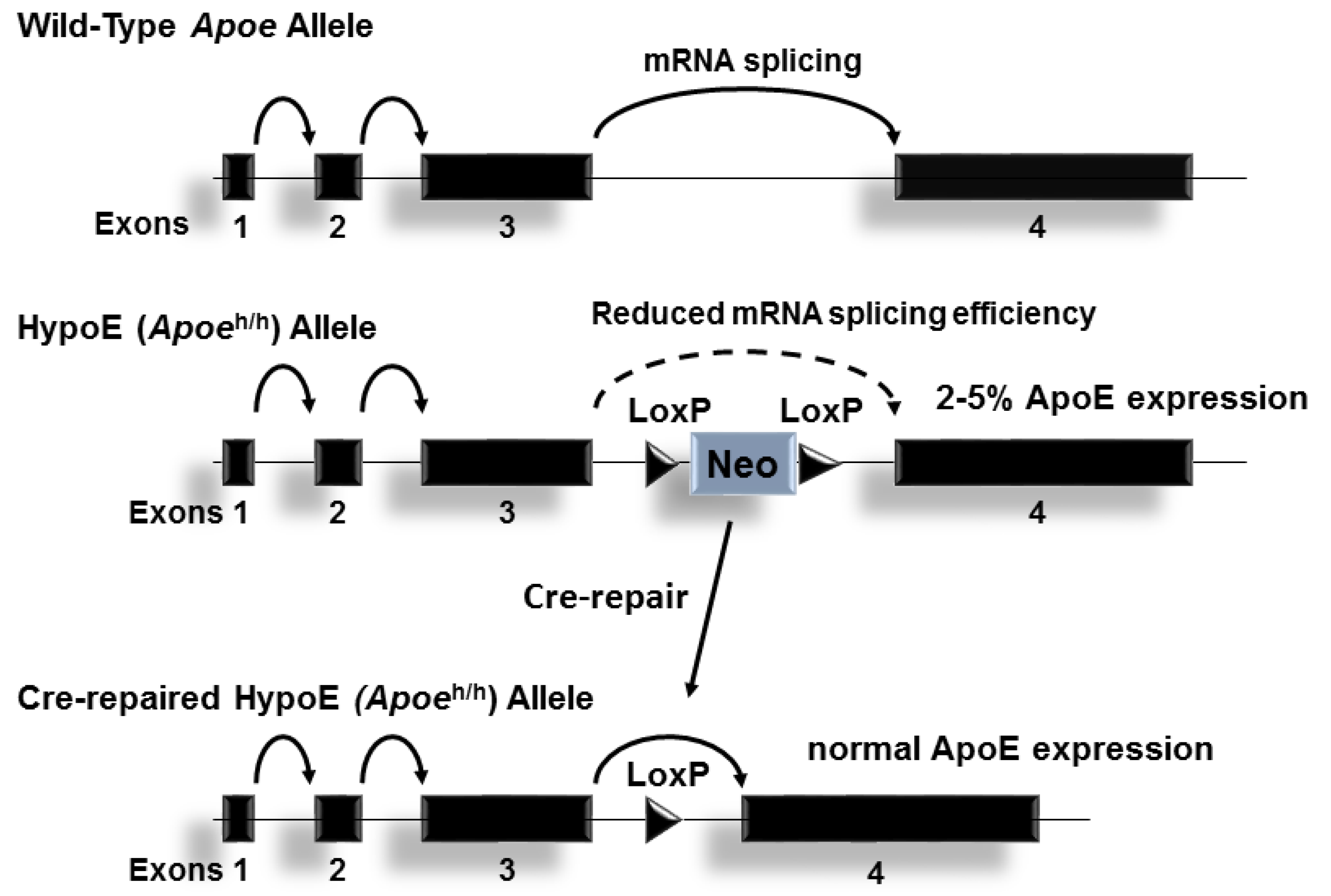
- Raffaï, R.L.; Weisgraber, K.H. Hypomorphic apolipoprotein e mice. A new model of conditional gene repair to examine apolipoprotein E-mediated metabolism. J. Biol. Chem. 2002, 277, 11064–11068. [Google Scholar] [CrossRef] [PubMed]
- Raffaï, R.L.; Hasty, A.H.; Wang, Y.; Mettler, S.E.; Sanan, D.A.; Linton, M.F.; Fazio, S.; Weisgraber, K.H. Hepatocyte-derived apoe is more effective than non-hepatocyte-derived apoe in remnant lipoprotein clearance. J. Biol. Chem. 2003, 278, 11670–11675. [Google Scholar] [CrossRef] [PubMed]
- Raffai, R.L.; Loeb, S.M.; Weisgraber, K.H. Apolipoprotein e promotes the regression of atherosclerosis independently of lowering plasma cholesterol levels. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Luk, F.S.; Kim, R.Y.; Olivas, V.R.; Kumar, N.; Posada, J.M.; Li, K.; Gaudreault, N.; Rapp, J.H.; Raffai, R.L. Inducible apoe gene repair in hypomorphic apoe mice deficient in the low-density lipoprotein receptor promotes atheroma stabilization with a human-like lipoprotein profile. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Picard, M.H.; Vasile, E.; Zhu, Y.; Raffai, R.L.; Weisgraber, K.H.; Krieger, M. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation 2005, 111, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kim, R.Y.; Imhof, I.; Honbo, N.; Luk, F.S.; Li, K.; Kumar, N.; Zhu, B.Q.; Eberle, D.; Ching, D.; et al. The immunosuppressant fty720 prolongs survival in a mouse model of diet-induced coronary atherosclerosis and myocardial infarction. J. Cardiovasc. Pharmacol. 2014, 63, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Luk, F.S.; Kim, R.Y.; Li, K.; Ching, D.; Wong, D.K.; Joshi, S.K.; Imhof, I.; Honbo, N.; Hoover, H.; Zhu, B.Q.; et al. Immunosuppression with fty720 reverses cardiac dysfunction in hypomorphic apoe mice deficient in Sr-Bi expression that survive myocardial infarction caused by coronary atherosclerosis. J. Cardiovasc. Pharmacol. 2016, 67, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Ng, Q.; Sanda, G.E.; Dey, A.K.; Teague, H.L.; Sorokin, A.V.; Dagur, P.K.; Silverman, J.I.; Harrington, C.L.; Rodante, J.A.; et al. Chronic skin inflammation accelerates macrophage cholesterol crystal formation and atherosclerosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Raffaï, R.L.; Dong, L.-M.; Farese, R.V., Jr.; Weisgraber, K.H. Introduction of human apolipoprotein e4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. USA 2001, 98, 11587–11591. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Kim, R.Y.; Luk, F.S.; de Mochel, N.S.; Gaudreault, N.; Olivas, V.R.; Kumar, N.; Posada, J.M.; Birkeland, A.C.; Rapp, J.H.; et al. Apolipoprotein E4 domain interaction accelerates diet-induced atherosclerosis in hypomorphic ARG-61 apoe mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Apoprotein e as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [PubMed]
- Hasty, A.H.; Linton, M.F.; Brandt, S.J.; Babaev, V.R.; Gleaves, L.A.; Fazio, S. Retroviral gene therapy in apoe-deficient mice. Apoe expression in the artery wall reduces early foam cell lesion formation. Circulation 1999, 99, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Fazio, S.; Babaev, V.R.; Burleigh, M.E.; Major, A.S.; Hasty, A.H.; Linton, M.F. Physiological expression of macrophage apoe in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J. Lipid Res. 2002, 43, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Bouchareychas, L.; Pirault, J.; Saint-Charles, F.; Deswaerte, V.; Le Roy, T.; Jessup, W.; Giral, P.; Le Goff, W.; Huby, T.; Gautier, E.L.; et al. Promoting macrophage survival delays progression of pre-existing atherosclerotic lesions through macrophage-derived apoe. Cardiovasc. Res. 2015, 108, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-abca1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of hdl, abca1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2009, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and b-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Stannard, A.K.; Riddell, D.R.; Sacre, S.M.; Tagalakis, A.D.; Langer, C.; von Eckardstein, A.; Cullen, P.; Athanasopoulos, T.; Dickson, G.; Owen, J.S. Cell-derived apolipoprotein E (Apoe) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J. Biol. Chem. 2001, 276, 46011–46016. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Graham, A.; Owen, J.S. Apolipoprotein e inhibits platelet aggregation through the l-arginine:Nitric oxide pathway. Implications for vascular disease. J. Biol. Chem. 1997, 272, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.J.; Reckless, J.; McKilligin, E. Apolipoprotein e modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 2004, 173, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Swertfeger, D.K.; Hui, D.Y. Apolipoprotein e receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J. Biol. Chem. 2001, 276, 25043–25048. [Google Scholar] [CrossRef] [PubMed]
- Swertfeger, D.K.; Bu, G.; Hui, D.Y. Low density lipoprotein receptor-related protein mediates apolipoprotein E inhibition of smooth muscle cell migration. J. Biol. Chem. 2002, 277, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, S.; Mahley, R.W.; Sanan, D.A.; Murata, J.; Newland, D.L.; Taylor, J.M.; Pitas, R.E. Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein e-null mice. J. Clin. Invest. 1995, 96, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Thorngate, F.E.; Rudel, L.L.; Walzem, R.L.; Williams, D.L. Low levels of extrahepatic nonmacrophage apoe inhibit atherosclerosis without correcting hypercholesterolemia in Apoe-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Wientgen, H.; Thorngate, F.E.; Omerhodzic, S.; Rolnitzky, L.; Fallon, J.T.; Williams, D.L.; Fisher, E.A. Subphysiologic apolipoprotein E (Apoe) plasma levels inhibit neointimal formation after arterial injury in apoe-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y.; Harmony, J.A.K.; Innerarity, T.L.; Mahley, R.W. Immunoregulatory plasma lipoproteins. Role of apoprotein e and apoprotein B. J. Biol. Chem. 1980, 255, 11775–11781. [Google Scholar] [PubMed]
- Pepe, M.G.; Curtiss, L.K. Apolipoprotein e is a biologically active constituent of the normal immunoregulatory lipoprotein, LDL-in. J. Immunol. 1986, 136, 3716–3723. [Google Scholar] [PubMed]
- Kelly, M.E.; Clay, M.A.; Mistry, M.J.; Hsieh-Li, H.-M.; Harmony, J.A.K. Apolipoprotein e inhibition of proliferation of mitogen-activated T lymphocytes: Production of interleukin 2 with reduced biological activity. Cell. Immunol. 1994, 159, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Tenger, C.; Zhou, X. Apolipoprotein e modulates immune activation by acting on the antigen-presenting cell. Immunology 2003, 109, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Middleton, M.; Pure, E.; Rader, D.J. Apolipoprotein e suppresses the type i inflammatory response in vivo. Circ. Res. 2005, 97, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Roselaar, S.E.; Daugherty, A. Apolipoprotein E-deficient mice have impaired innate immune responses to listeria monocytogenes in vivo. J. Lipid Res. 1998, 39, 1740–1743. [Google Scholar] [PubMed]
- De Bont, N.; Netea, M.G.; Demacker, P.N.M.; Kullberg, B.J.; van der Meer, J.W.M.; Stalenhoef, A.F.H. Apolipoprotein e-deficient mice have an impaired immune response to klebsiella pneumoniae. Eur. J. Clin. Invest. 2000, 30, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Van Oosten, M.; Rensen, P.C.; Van Amersfoort, E.S.; Van Eck, M.; Van Dam, A.M.; Breve, J.J.; Vogel, T.; Panet, A.; Van Berkel, T.J.; Kuiper, J. Apolipoprotein E protects against bacterial lipopolysaccharide-induced lethality. A new therapeutic approach to treat gram-negative sepsis. J. Biol. Chem. 2001, 276, 8820–8824. [Google Scholar] [CrossRef] [PubMed]
- Kattan, O.M.; Kasravi, F.B.; Elford, E.L.; Schell, M.T.; Harris, H.W. Apolipoprotein E-mediated immune regulation in sepsis. J. Immunol. 2008, 181, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Li, Z.; Mazzone, T. Tumor necrosis factor-a modulates monocyte/macrophage apoprotein e gene expression. J. Clin. Invest. 1995, 96, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.; Mackman, N.; Curtiss, L.K. Interferon-g inhibits macrophage apolipoprotein E production by posttranslational mechanisms. J. Clin. Investig. 1993, 91, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Garlanda, C.; Locati, M. Macrophage diversity and polarization in atherosclerosis: A question of balance. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Adamson, S.; Leitinger, N. Phenotypic modulation of macrophages in response to plaque lipids. Curr. Opin. Lipidol. 2011, 22, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via NRF2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, J.E.; Parathath, S.; Rong, J.X.; Mick, S.L.; Vengrenyuk, Y.; Grauer, L.; Young, S.G.; Fisher, E.A. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011, 123, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, D.; Bock, H.H.; Engel, T.; Telgmann, R.; Muller-Tidow, C.; Varga, G.; Bot, M.; Herz, J.; Robenek, H.; von Eckardstein, A.; et al. Apolipoprotein e induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Vengrenyuk, Y.; Ramsey, S.A.; Vila, N.R.; Girgis, N.M.; Liu, J.; Gusarova, V.; Gromada, J.; Weinstock, A.; Moore, K.J.; et al. Inflammatory ly6chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 2017, 127, 2904–2915. [Google Scholar] [CrossRef] [PubMed]
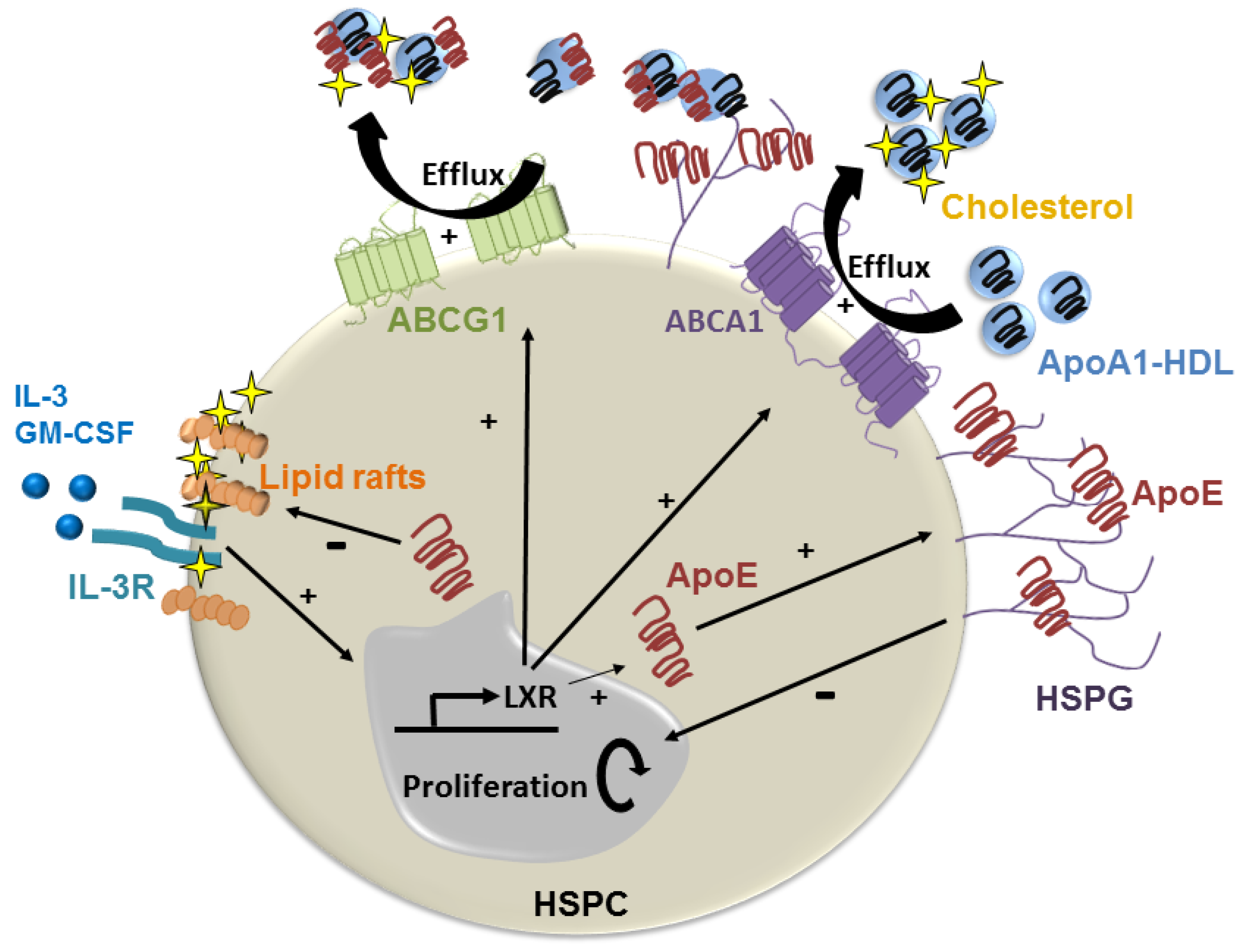
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and hdl suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Huang, Z.H.; Mazzone, T. Interaction with proteoglycans enhances the sterol efflux produced by endogenous expression of macrophage apoe. J. Lipid Res. 2001, 42, 1125–1133. [Google Scholar] [PubMed]
- Rosenfeld, M.E.; Butler, S.; Ord, V.A.; Lipton, B.A.; Dyer, C.A.; Curtiss, L.K.; Palinski, W.; Witztum, J.L. Abundant expression of apoprotein e by macrophages in human and rabbit atherosclerotic lesions. Arterioscler. Thromb. 1993, 13, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, S.; Wara, A.K.; Icli, B.; Shvartz, E.; Tesmenitsky, Y.; Belkin, N.; Li, D.; Blackwell, T.S.; Sukhova, G.K.; et al. Systemic delivery of microrna-181B inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein e-deficient mice. Circ. Res. 2014, 114, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing BCL6 in macrophages. J. Clin. Invesigt. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.W.; Moore, K.J. Microrna regulation of atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. Nf-kappab dysregulation in microrna-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. Mir-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. Nf-kappab-dependent induction of microRNA mir-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Etzrodt, M.; Cortez-Retamozo, V.; Newton, A.; Zhao, J.; Ng, A.; Wildgruber, M.; Romero, P.; Wurdinger, T.; Xavier, R.; Geissmann, F.; et al. Regulation of monocyte functional heterogeneity by mir-146a and RELB. Cell Rep. 2012, 1, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.L.; Rao, D.S.; O’Connell, R.M.; Garcia-Flores, Y.; Baltimore, D. MicroRNA-146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. eLife 2013, 2, e00537. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in es cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef] [PubMed]
- De Bont, N.; Netea, M.G.; Demacker, P.N.; Verschueren, I.; Kullberg, B.J.; van Dijk, K.W.; van der Meer, J.W.; Stalenhoef, A.F. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and klebsiella pneumoniae infection. J. Lipid Res. 1999, 40, 680–685. [Google Scholar] [CrossRef]
- Cheng, H.S.; Sivachandran, N.; Lau, A.; Boudreau, E.; Zhao, J.L.; Baltimore, D.; Delgado-Olguin, P.; Cybulsky, M.I.; Fish, J.E. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol. Med. 2013, 5, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; Chen, L.; Zhang, P.; Chen, H.; Liu, Y.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional apolipoprotein e. Cell Death Dis. 2018, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouchareychas, L.; Raffai, R.L. Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation. J. Cardiovasc. Dev. Dis. 2018, 5, 30. https://doi.org/10.3390/jcdd5020030
Bouchareychas L, Raffai RL. Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation. Journal of Cardiovascular Development and Disease. 2018; 5(2):30. https://doi.org/10.3390/jcdd5020030
Chicago/Turabian StyleBouchareychas, Laura, and Robert L. Raffai. 2018. "Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation" Journal of Cardiovascular Development and Disease 5, no. 2: 30. https://doi.org/10.3390/jcdd5020030
APA StyleBouchareychas, L., & Raffai, R. L. (2018). Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation. Journal of Cardiovascular Development and Disease, 5(2), 30. https://doi.org/10.3390/jcdd5020030